Лучевые признаки синдрома Холт-Орама у плода
Обновлено: 28.04.2024
Цель: демонстрация клинического течения и результатов лечения синдрома Холта-Орама.
Синдром Холта-Орама (предсердно-пальцевая дисплазия, синдром "рука-сердце") впервые описали M.Holt и S.Oram в 1960 г. Частота встречаемости в популяции неизвестна. Соотношение полов при этой патологии составляет 1:1. Тип наследования - аутосомно-доминантный с различной экспрессивностью. Минимальные диагностические признаки: пороки развития верхних конечностей и врожденные дисплазии сердца.
В 85% случаев при данном синдроме выявляются врожденные пороки сердца: дефект межпредсердной перегородки (наиболее часто), дефект межжелудочковой перегородки, открытый артериальный проток, коартация аорты, стеноз легочной артерии. Часто имеется пролапс митрального клапана.
С рождения обнаруживаются нарушения ритма и проводимости сердца. Пороки развития верхних конечностей варьируют от изменений 1 пальца кисти и гипоплазии лучевой кости до фокомелии. К ним относятся поворот лопатки в сторону аксилярной линии грудной клетки, расширение наружного конца головки ключицы, аномальная форма клювовидного отростка лопатки, изменение формы ладьевидной кости, отсутствие, гипертрофия или порочное расположение большого пальца с недостаточным противопоставлением, синдактилия, клинодактилия, воронкообразная грудная клетка.
В редких случаях описывают другие стигмы эмбриогенеза - гипертелоризм, отсутствие большой грудной мышцы, расщелина неба. Биохимические нарушения - задержка азота, повышение уровня мочевины в крови. Нормохромная анемия, спинальная гипоплазия псевдо-Фанкони. Интеллект, как правило, сохранен. Этиопатогенез синдрома - гистодисплазия, нарушение формирования первичного сердца и верхних конечностей на 5-й неделе эмбрионального развития.
Эволюция синдрома заключается в прогрессировании и декомпенсация сердечного порока, развитии нарушений сердечного ритма (СР). Кроме того, отмечается снижение клеточного иммунитета и инфекционные осложнения. Приводим собственные наблюдения клинического течения данного синдрома у двух пациентов.
Один из них, Владислав З., 24 лет, находился на отделении лечения нарушений ритма сердца и электрокардиостимуляции (ЭКС) Клинического центра передовых медицинских технологий с диагнозом: Синдром Холта- -Орама. Врожденная аномалия сердца - синдром бинодальной слабости.
Пациент жаловался на вялость, слабость, головные боли, эпизоды головокружения. Синкопальных состояний не было. Из раннего анамнеза известно, что ребенок от 2-й беременности, сопровождавшейся токсикозом 1-й и 2-й половины и патологической прибавкой массы тела во 2-й половине. Первая беременность в 1965 г. была прервана по причине болезни матери.
На момент беременности матери было 34 года, профессия - химик-технолог, работала на химическом производстве с профессиональной вредностью (имины, фторорганика). За период беременности мать дважды болела ОРВИ, до беременности в течение 12 лет страдала туберкулезным хориоретинитом. Из-за болезни матери ребенок родился в инфекционной больнице путем кесарева сечения с весом 3050 г, длиной тела 50 см. Через месяц после родов был отмечен систолический шум в области сердца, неправильное развитие пальцев левой кисти.
В возрасте 6 месяцев ребенок находился на стационарном лечении в инфекционной больнице с диагнозом: ОРЗ, двусторонняя пневмония, врожденный порок сердца (?), гипотрофия 1-2 степени, рахит 1-2 степени, стафилококковый энтерит, лямблиоз кишечника. Врожденная правосторонняя паховая грыжа. Водянка правого яичка. Врожденная двусторонняя лучевая косорукость.
В 1 год отмечено отставание в весе, психоэмоциональное развитие по возрасту. В 4 года выполнена корригирующая операция по поводу врожденного порока развития кистей - трехфалангового большого пальца, клинодактилии 1 пальца левой кисти (удаление добавочной средней фаланги, устранение клинодактилии). Осталась гипоплазия мышц тенара, резкое нарушение супинации костей без рентгенологически определяемого синостоза, воронкообразная грудная клетка.
В анамнезе ребенка частые ОРВИ, скарлатина, краснуха, эпидпаротит, хронический тонзиллит с частыми обострениями, постоянное отставание в физическом развитии. В 11 лет оперирован по поводу острого аппендицита.
На диспансерном учете у кардиоревматолога состоял с рождения. Первая ЭКГ в возрасте 10 мес. - синусовый ритм, брадикардия 105 уд/мин (Р-Р = 570 мс, P-Q = 160 мc), тенденция к отклонению электрической оси сердца вправо. Неполная блокада правой ножки пучка Гиса.
В возрасте 2,5 лет на ЭКГ регистрировалась синусовая брадиаритмия с частотой сердечных сокращений 77 уд/мин, небольшое замедление атриовентрикулярной проводимости (P-Q = 180 мс). С 3,5 лет - выраженная синусовая брадиаритмия от 83 до 53 в мин. В 7 лет наблюдалась миграция водителя ритма по предсердиям, частота ритма 46 уд/мин.
В 8 лет отмечено появление изоритмической АВ диссоциации, а в 9 лет впервые поставлен диагноз синдрома слабости синусового узла (брадикардия 45 уд/мин, постоянная интерферирующая АВ диссоциация, предсердная экстрасистолия). При дальнейшем наблюдении ЭКГ без существенной динамики.
В возрасте 15 лет после консультации в генетическом центре ребенку был поставлен диагноз: синдром Холта-Орама. При эхокардиографическом исследовании выявлены пролапсы митрального, трикуспидального клапанов и клапана легочной артерии 1 степени.
При рентгенографии грудной клетки в возрасте 9 лет выявлено умеренное усиление легочного рисунка в корневых зонах, расширение сердца в поперечнике за счет всех сегментов (КТИ = 57 при норме до 50). В 16 лет отмечено усиление легочного рисунка за счет гиперволемии, расширение легочного ствола, увеличение правых камер сердца и левого желудочка.
В возрасте 22 лет при Холтеровском суточном мониторировании отмечена выраженная синусовая брадиаритмия, изоритмическая АВ диссоциация с частотой ритма до 28 уд/мин, паузы от 1240 до 3338 мс за счет СА блокады 2 степени 2 типа, 2:1, 3:1 и остановки синусового узла. За время наблюдения зафиксировано 3340 пауз (в среднем 182 в час). Обращало на себя внимание отсутствие выскальзывающих комплексов из центров автоматизма 2 порядка в моменты асистолии. Периодически регистрировалась АВ блокада 1 типа (PQ = 240 мс).
При ЧП ЭФИ максимальное ВВФСУ = 1800 мс (норма до 1540 мс), КВВФСУ = 800 мс (норма до 540 мс), антероградное АВ проведение на нижней границе нормы (точка Венкебаха АВ соединения = 130 имп/мин). Постстимуляционные паузы достигали 2200 мс. После введения атропина максимальная частота ритма составила всего 85 в минуту (при норме не менее 105 в мин). Заключение: Синдром слабости синусового узла (СССУ). Формирование синдрома бинодальной слабости.
Пациенту была предложена операция: имплантация постоянного электрокардиостимулятора (ПЭКС). В отделении ему был имплантирован ПЭКС фирмы Intermedics "Relay" в режиме DDDR. Импортный физиологический электрокардиостимулятор был выделен для пациента по распоряжению Президента РФ.
Улучшение самочувствия пациент отметил уже на второй день после операции. Исчезли слабость, вялость, головокружение. Установлены физиологические параметры стимуляции при удовлетворительных пороговых значениях. При контрольном посещении через три и шесть месяцев после выписки из стационара жалоб у пациента нет, отмечает улучшение аппетита, настроения, повышение толерантности к нагрузкам (по собственному выражению пациента, стал "летать"). В настоящее время закончил аспирантуру и завершает работу над кандидатской диссертацией.
Другая пациентка: Катя Б., 6 лет. Признаками синдрома Холта-Орама являлись врожденная патология обеих кистей, недоразвитие лучевых костей, дефект межпредсердной перегородки, оперированный в возрасте 1,5 лет, нарушения ритма сердца в виде синдрома слабости синусового узла (брадикардия, эпизоды изоритмической АВ-диссоциации и замещающего АВ-ритма, АВ узловая экстрасистолия, отсутствие прироста частоты сердечных сокращений на физическую нагрузку и введение атропина).
По совокупности данных обследования были определены показания для имплантации искусственного водителя ритма. Девочке имплантирован физиологический однокамерный ПЭКС фирмы Pacesetter "Regency SR", в который включен частотно- адаптивный режим VVIR. Параметры электростимуляции удовлетворительные, у девочки улучшился аппетит, уменьшились жалобы. Теперь ей предстоит хирургическая коррекция кистей, для которой сейчас отсутствуют противопоказания со стороны кардиальной патологии.
В заключение следует отметить, что брадиаритмия у пациентов с синдромом Холта-Орама имеет тенденцию к прогрессивному ухудшению с формированием СССУ, а своевременная имплантация физиологической ПЭКС-системы позволяет нормализовать клиническую картину, гемодинамические параметры и значительно улучшить качество жизни.
ЛИТЕРАТУРА
1. Справочник клинических симптомов и синдромов. Под ред. И.Р.Лазовскис. - М., Мед., 1981 г.
2. Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование. Л.:Медицина, 1987. - с. 227-228.
3. Парщикова Л.С., Рейдерман М.И. Синдром Холта-Орама //Ортопедия, травматология. - 1976. - N 10, с. 69-70.
5. Chang C.H. Holt-Oram Syndrome.//Radiology. - 1967. - v. 88. - p. 479-483.
6. Holt M., Oram S. Familian heart disease with skeletal malformations // Brit. Heart J. - 1960. - N 2. - p. 236.
7. Martin Cl., Badin J., San Juan B., Hehunstere J. Un cas de Syndrome de Holt-Oram // Arch Franc Ped. - 1970. - N 8. - p. 865.
8. Gladstone I.A., Subert V.P. Holt-Oram syndrome: Penetrance of the gene and lack of maternal effect // Clin Genet. - 1982. - V. 21. - p. 99-103.
Синдром Холта-Орама ( Предсердно-пальцевая дисплазия , Синдром «рука-сердце» )
Синдром Холта-Орама — это редкая наследственная патология, которая характеризуется морфологическими аномалиями верхних конечностей, разнообразными врожденными сердечными пороками. Возникает вследствие генной мутации TBX5, наследуется по аутосомно-доминантному типу. Для диагностики синдрома Холта-Орама назначается рентгенологическое исследование кисти, эхокардиография и электрокардиография, наличие мутации подтверждается с помощью генетического тестирования. Лечение включает хирургические операции по устранению сердечных пороков, помощь ортопедов-травматологов при тяжелых деформациях опорно-двигательного аппарата.
МКБ-10
Общие сведения
Синдром Холта-Орама имеет дополнительные наименования в медицинской литературе: синдром «рука-сердце», предсердно-пальцевая дисплазия. Типичные клинические проявления патологии впервые описали в 1960 г. британские кардиологи Мери Холт и Сэмюэл Орам, в честь которых названо заболевание. Точные генетические причины развития скелетных и сердечных аномалий были установлены в 1997 г. Болезнь Холта-Орама относится к орфанным патологиям, встречается с частотой 1 случай на 100 тыс. населения, не имеет половых различий.
Причины
Заболевание вызвано мутациями в гене TBX5, который располагается на длинном плече 12-й хромосомы в локусе 12q24.21. Патология имеет аутосомно-доминантный тип наследования, однако нередки случаи спонтанного возникновения мутации у ребенка, который имеет абсолютно здоровых родителей. Генная мутация имеет разную степень экспрессивности, что обуславливает вариабельность клинической картины синдрома.
Патогенез
Типичные морфологические изменения при болезни Холта-Орама связаны с отсутствием или недостаточным синтезом транскрипционного фактора Tbx5, принадлежащего к семейству T-box. Он регулирует многие процессы формирования соединительной ткани при внутриутробном развитии, включая развитие межпредсердных и межжелудочковых перегородок, проводящей системы сердца, мышц и сухожилий.
При недостаточном уровне протеина Tbx5 обычно нарушается строение перегородки сердца, аортального клапана, именно в этих зонах возникает до 95% всех врожденных пороков у страдающих синдромом Холта-Орама. Также наличие транскрипционного фактора является обязательным условием для дифференцировки кардиомиоцитов проводящей системы сердца, поэтому патология часто сопровождается нарушением ритма.
Симптомы
Основным признаком синдрома Холта-Орама служат аномалии строения верхней конечности, которые выявляются у 100% больных. Они преимущественно проявляются изолированной гипоплазией тенара — ладонного возвышения в основании большого пальца, появлением третьей фаланги на большом пальце, лево-правой асимметрией. В тяжелых случаях синдрома возникают грубые инвалидизирующие пороки развития — фокомелия (тюленеобразные конечности).
Вторым по частоте симптомом, который наблюдается у 85% пациентов являются врожденные пороки сердца. Чаще всего у больных обнаруживаются дефект межпредсердной перегородки (44,4%), дефект межжелудочковой перегородки (29,4%). Намного реже встречаются грубые сердечные аномалии — тетрада Фалло, синдром гипоплазии левых отделов сердца, коарктация аорты.
Третьим по распространенности проявлением синдрома Холта-Орама являются нарушения ритма и проводимости, которые регистрируются в 40% случаев. Они представлены синусовой брадикардией, блокадой правой ножки пучка Гиса, атриовентрикулярной диссоциацией или синдромом слабости синусового узла. В редких случаях определяются стигмы эмбриогенеза: гипертелоризм, расщелина неба, отсутствие большой грудной мышцы.
Осложнения
Пациенты с синдромом Холта-Орама зачастую отстают в росте и физическом развитии с раннего детства, при этом психическое развития происходит согласно возрасту. Если не проводится кардиохирургическая коррекция, на фоне сердечных пороков наблюдаются нарушения кровообращения, вплоть до тяжелой сердечной недостаточности. При длительном существовании синдрома снижается клеточный иммунитет, из-за возникают частые ОРЗ, пневмонии, отиты и синуситы.
Диагностика
Поскольку первые признаки синдрома заметны уже в раннем детском возрасте, обследованием ребенка занимается педиатр, по показаниям к диагностике привлекают детского кардиохирурга, генетика, ортопеда-травматолога. Заподозрить диагноз удается по наличию специфических деформаций верхних конечностей. Для подтверждения синдрома Холта-Орама назначаются следующие диагностические методы:
- Рентгенография кисти. На рентгеновском снимке определяют гипоплазию тенара, появление у большого пальца одной или обеих рук третьей фаланги, недостаточное противопоставление большого пальца. Изредка выявляются более тяжелых деформации костей руки с вовлечением в процесс пояса верхних конечностей.
- Эхокардиография. Ультразвуковое исследование сердца — ведущий метод диагностики врожденных аномалий, характерных для синдрома. При осмотре удается визуализировать структурные изменения органа, оценить его функциональные возможности. Для уточнения диагноза применяется рентгенография ОГК.
- Электрокардиография. Метод позволяет вовремя обнаружить признаки синусовой аритмии периодического и апериодического типа, синоатриальных блокад, синусовой тахикардии, предсердной экстрасистолии, других типичных нарушений ритма. В ряде случаев для более точной диагностики рекомендуется суточное мониторирование ЭКГ.
- Генетическое тестирование. Поиск специфической мутации путем секвенирования экзона — основной метод подтверждения диагноза, который используется в сомнительных случаях, в рамках научных исследований. В рутинной практике, как правило, достаточно стандартных клинических и инструментальных методов.
Лечение синдрома Холта-Орама
Терапевтические мероприятия подбираются с учетом характера и степени клинических проявлений синдрома. Основу лечения составляет помощь кардиохирургов для ликвидации врожденных пороков сердца, позволяющая устранить расстройства кровообращения, обеспечить больным высокую продолжительность и уровень жизни. В зависимости от типа порока хирургическое вмешательство производится в разном возрасте:
- При дефекте межжелудочковой перегородки оптимальный возраст коррекции составляет от 1 до 2-2,5 лет, что позволяет избежать тяжелых осложнений, обеспечить нормально физическое развитие ребенка.
- При дефекте межпредсердной перегородки наилучшие результаты коррекции достигаются при выполнении операции в дошкольном возрасте (3-6 лет).
- При тяжелых пороках (например, тетраде Фалло) кардиохирургическая операция проводится в 4-6 месяцев, а при наличии одышечно-цианотических приступов — еще раньше.
- При жизнеугрожающих аритмиях показана имплантация электрокардиостимулятора, если медикаментозные методы лечения не дают эффекта.
Помимо кардиохирургической помощи, больному могут потребоваться реконструктивные операции для уменьшения степени дефекта верхней конечностей, повышения ее функциональных возможностей. В течение всей жизни пациент с болезнью Холта-Орама пребывает под диспансерным наблюдением кардиолога, ортопеда, генетика.
Прогноз и профилактика
Прогноз определяется степенью тяжести поражения сердца: негрубые пороки можно полностью устранить при кардиохирургической операции, однако изредка встречаются тяжелые аномалии сердца с высоким риском летального исхода. Учитывая спонтанный характер мутации, меры первичной профилактики синдрома не разработаны. Вторичная профилактика включает своевременную диагностику, кардиохирургическую помощь в полном объеме.
1. Множественные мальформации сердца у пациента с синдромом Холта-Орама/ И.А. Сойнов, Д.А. Дульцева, А.В. Лейкхеман, А.Н. Архипов// Российский вестник перинатологии и педиатрии. — 2020. — №5.
2. Клинический случай: синдром Холта-Орама/ А.С. Хайбуллина // 50-я ежегодная научно-практическая конференция студентов и молодых учёных по итогам летней производственной практики. — 2017.
3. Синдром Холта-Орама/ О.Л. Цимбалиста, Н.Н. Фоменко, Т.М. Мельник, Е.Д. Шустакевич// Здоровье ребенка. — 2010. — №6.
4. Клинический случай синдрома Холта-Орама у ребенка / С.Я. Волгина, Н.И. Клейменова // Казанский медицинский журнал. — 2009. — №6.
Синдром Эдвардса
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причины синдрома Эдвардса
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
Внешний облик детей дополняется характерными для синдрома Эдвардса деформациями скелета - скрещенными пальцами кистей, укороченной грудиной, аномалиями ребер, врожденным вывихом бедра, косолапостью, «стопой-качалкой», синдактилией стоп и пр. У многих детей имеются гемангиомы и папилломы кожи.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Пороки развития центральной нервной системы характеризуются наличием микроцефалии, менингомиелоцеле, гидроцефалии, аномалии Арнольда-Киари, кист арахноидального сплетения, гипоплазии мозжечка и мозолистого тела. У всех выживших детей с синдромом Эдвардса имеются интеллектуальные нарушения - олигофрения в степени глубокой имбецильности или идиотии.
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Лучевые признаки синдрома Холт-Орама у плода
УЗИ, МРТ при синдроме Гольденхара у плода
а) Терминология:
1. Синонимы:
• Окуло-аурикуло-вертебральная дисплазия (ОАВД):
о В настоящее время данный термин считается наиболее точным
2. Определения:
• Нарушение развития производных первой и второй жаберных дуг
• Единое мнение в отношении минимально необходимого количества диагностических критериев отсутствует:
о Минимально необходимыми диагностическими критериями считаются микротия (уменьшение размеров наружного уха) или гемифациальная микросомия + пороки развития органа слуха:
- Строгое определение микротии включает отсутствие наружного слухового прохода
о Достаточным критерием диагностики может служить изолированная гемифациальная микросомия в сочетании с положительным семейным анамнезом ОАВД
о В 50% случаев обнаруживают сопутствующую патологию сердца, головного мозга, легких или мочеполовой системы

(Слева) 3D УЗИ плода с подозрением на расщелину верхней губы. В действительности расщелина отсутствует, однако отмечается асимметрия лица: левая глазница уменьшена, кончик носа деформирован.
(Справа) Асимметрия ушных раковин: правая сформирована нормально, левая уменьшена и уплощена. Пороки развития лица и ушных раковин при синдроме Гольденхара встречаются довольно часто. В данном случае также обнаружена и подтверждена с помощью МРТ АМТ (отсутствие ППП).
б) Лучевая диагностика:
1. МРТ при синдроме Гольденхара у плода:
• Дополняет исследование органа зрения (например, позволяет диагностировать колобому)
• МРТ в режиме Т2-ВИ позволяет исследовать строение лица, если поверхностная реконструкция с помощью 3D УЗИ невозможна
• Позволяет уточнить характер аномалий головного мозга или позвоночника
2. УЗИ при синдроме Гольденхара у плода:
• Пренатальная диагностика основана на обнаружении пороков развития лица (52,4%), особенно на фоне множественных сочетанных аномалий:
о Пороки сердца (19%):
- Тетрада Фалло, транспозиция магистральных сосудов, дефекты перегородок, транспозиция сердца, декстрокардия, аномалии дуги аорты
о Аномалии головного мозга (47,6%):
- Микроцефалия, дефекты заращения нервной трубки, патология мозолистого тела, ГПЭ
о Аномалии развития мочеполовой системы:
- Односторонняя агенезия почки, ДМП
о Дефекты радиального луча
3. Рекомендации по лучевой диагностике:
• Предпочтительный метод исследования:
о 3D УЗИ позволяет оценить строение наружного уха и асимметрию лица
в) Дифференциальная диагностика синдрома Гольденхара у плода:
1. Синдром CHARGE:
• Характерная форма ушных раковин, патология полукружных каналов
2. Синдром Тричера Коллинза:
• Выраженная микрогнатия, антимонголоидный разрез глаз
(Слева) МРТ плода с подозрением на синдром каудальной регрессии, Т2-ВИ, сагиттальная плоскость. УЗИ оказалось невозможным по причине ожирения матери. Позвоночник прерывается на уровне L4, размеры таза уменьшены. На клинической фотографии новорожденного определяются укорочение туловища, патологическое положение ног и кожные лоскуты кпереди от ушной раковины. Окончательный диагноз - ОАВД.
(Справа) Посмертная рентгенография. Определяются асимметрия глазниц и заращение наружного слухового канала. Левая половина лица уменьшена, присутствует односторонний микрофтальм.
г) Патологоанатомические особенности:
1. Общие сведения:
• Гипоплазия верхней ± нижней челюсти, асимметрия скуловых дуг, скуловых и височных костей, наружного и среднего уха, мышц лица:
о ± аномалии развития других производных жаберных дуг (органа зрения, позвоночника, верхней части сердца)
о ± аномалии развития органов, происходящих не от жаберных дуг (например, почек)
• Термином «гемифациальный» обозначают одностороннюю локализацию поражений, однако в большинстве случаев наблюдают двустороннее асимметричное расположение
2. Генетические факторы:
• Заболевание чаще всего возникает случайным образом, однако описаны случаи аутосомно-доминантного и -рецессивного наследования
• Возникает в результате различных хромосомных аберраций и геномной неустойчивости
• Эмпирически установленный риск повторного возникновения при нормальном наборе хромосом и отрицатедьном семейном анамнезе составляет 2-3%
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о В большинстве случаев присутствует асимметрия лица:
- Аномалии чаще всего односторонние, однако возможна двусторонняя локализация с преимущественным поражением правой стороны
• Другие субъективные и объективные симптомы:
о Асимметричные пороки развития органа слуха
о Орофациальные расщелины (редко), макростомия (часто)
о Поражения органа зрения: эпибульбарный дермоид (нередко определяется в постнатальном периоде), микр-офтальм
о Аномалии развития позвоночника
2. Демографические особенности:
• Эпидемиология:
о 1:3000-5000 родов
3. Естественное течение и прогноз:
• Прогноз зависит от сопутствующих пороков развития
• Тугоухость, затруднения при грудном вскармливании, дефекты речи, расстройства сна
• Внешность ребенка оказывает негативное влияние на психологическое состояние самого ребенка и членов его семьи
4. Лечение синдрома Гольденхара у плода:
• Поддерживающая терапия во время беременности
• Тщательное аудиологическое обследование новорожденного
• Для коррекции асимметрии и воссоздания более привычной внешности показана челюстно-лицевая хирургия
е) Особенности диагностики:
1. Важно знать:
• Наиболее тяжелым течением характеризуются случаи заболевания, диагностированные пренатально
2. Признаки, учитываемые при интерпретации изображений:
• Фенотипические проявления широко варьируют, пренатальная диагностика может быть невозможна даже в случае положительного семейного анамнеза
Редактор: Искандер Милевски. Дата обновления публикации: 18.11.2021
УЗИ при синдроме Холт-Орама у плода
а) Определения:
• Синдром Холт-Орама, также известный как «синдром рука-сердце», характеризуется аномалиями развития верхней конечности и сердца
1. Общие сведения:
• Критерии диагностики:
о Дефект радиального луча у плода с положительным семейным анамнезом в отношении синдрома Холт-Орама
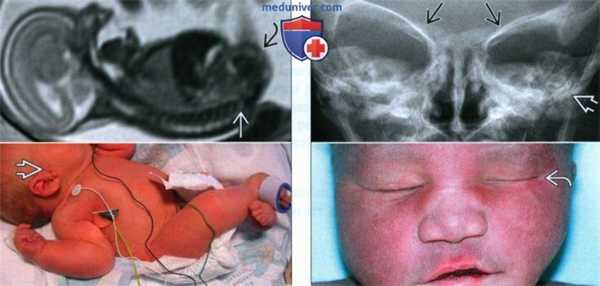
(Слева) УЗИ плода в 19 нед. Синдром Холт-Орама. На кисти определяются 4 пальца, I палец отсутствует. Отмечается клинодактилия V пальца.
(Справа) Тот же случай. Клиническая фотография кисти новорожденного. Медиальная девиация кисти обусловлена отсутствием лучевой кости Кроме того, плечевая и локтевая кости гипоплазированы. I палец отсутствует, присутствуют только 4 пальца. Клинодактилия V пальца совпадает с более ранней находкой по УЗИ. (Слева) УЗИ плода во II триместре. Синдром Холт-Орама. Определяется дефект радиального луча: лучевая кость гипоплазирована, кисть медиально отклонена. Изначально по результатам пренатальной лучевой диагностики предполагалось отсутствие I пальца кисти, однако после рождения ребенка установлена гипоплазия пальца.
(Справа) Четырехкамерный срез сердца плода в III триместре. Визуализируется небольшой ДМЖП. Самым частым пороком сердца, встречающимся при синдроме Холт-Орама, является ДМПП, обнаружить который до родов особенно сложно. (Слева) УЗИ верхней конечности плода в 26 нед. Синдром Холт-Орама. Отмечаются выраженная гипоплазия лучевой кости медиальная девиация кисти и олигодактилия. I палец отсутствует.
(Справа) Клиническая фотография. Рука женщины с тяжелой формой синдрома Холт-Орама. Присутствуют только 4 пальца, определяются камптодактилия, а также выраженное укорочение руки вследствие аплазии лучевой и гипоплазии плечевой кости.
2. УЗИ при синдроме Холт-Орама у плода:
• Дефекты радиального луча, определяемые с I триместра:
о Первая и наиболее явная патологическая находка у плода с положительным семейным анамнезом
• Выраженность дефицита радиального луча варьирует, обычно присутствует асимметрия
• Пороки развития нижних конечностей (реже)
• Пороки сердца:
о Самый частый порок сердца - ДМПП; во внутриутробном периоде диагностируется редко
о ДМЖП встречается реже
3. Рекомендации по лучевой диагностике:
• Советы по проведению исследования:
о 3D/4D УЗИ позволяет более детально исследовать дефекты конечностей
о ЭхоКГ показана всем плодам из группы высокого риска, в том числе при отсутствии явных дефектов конечностей
4. Рентгенография при синдроме Холт-Орама у плода:
• Поражение верхних конечностей включает различную степень гипоплазии или аплазию лучевой кости, костей запястья и I пальца
в) Дифференциальная диагностика синдрома Холт-Орама у плода:
1. Дефекты радиального луча, изолированные или в составе синдромов:
• Анемия Фанкони:
о Аплазия лучевой кости о Различные варианты поражения I пальца
• Тромбоцитопения с отсутствием лучевой кости (TAR-синдром):
о I палец присутствует всегда о Аплазия лучевой кости
• Ассоциация VACTERL:
о Аномалии развития позвонков, атрезия ануса, пороки сердца, трахеопищеводный свищ, АП, агенезия почек, различные аномалии развития конечностей, в том числе дефекты радиального луча
• Изолированная гипоплазия или трехфалангизм I пальца кисти
2. Изолированный ДМПП:
• Диагностика во внутриутробном периоде затруднена
(Слева) УЗИ кисти плода в конце I триместра. Синдром Холт-Орама, дефект радиального луча верхней конечности. Определяются только 4 пальца, I палец отсутствует.
(Справа) УЗИ плода во II триместре. Синдром Холт-Орама. Отмечаются умеренная гипоплазия лучевой кости, медиальная девиация, а также выраженная гипоплазия кисти. Локтевая кость умеренно искривлена. (Слева) УЗИ плода в 12 нед. Беременная страдает синдромом Холт-Орама. Визуализируются признаки дефекта радиального луча, а также косорукость. Указанные патологические находки, а также отягощенный анамнез матери позволяют заподозрить у плода синдром Холт-Орама.
(Справа) Тот же случай. Клиническая фотография. Диагноз синдрома Холт-Орама подтверждается при осмотре новорожденного. Определяются двусторонние дефекты радиальных лучей (поражение слева более тяжелое, чем справа). Большие пальцы отсутствуют на обеих кистях. По результатам ЭхоКГ плода и новорожденного нарушения не обнаружены. (Слева) Тот же случай. Клиническая фотография кисти, принадлежащей матери новорожденного. Отмечаются проксимально расположенный гипоплазированный I палец и шрам от хирургической операции на сухожилиях
(Справа) Клиническая фотография обеих кистей женщины. Определяется трехфалангизм I пальца правой кисти. Пациентка считает эту руку здоровой. Левая рука сравнительно меньших размеров супинация кисти ограничена. Также в детстве у женщины был диагностирован ДМЖП, впоследствии устраненный с помощью хирургического лечения.
г) Патологоанатомические особенности. Общие сведения:
• Этиология:
о Мутации гена, кодирующего транскрипционный фактор ТВХ5 (12q24.21):
- Мутации приводят к функциональной гаплонедостаточ-ности с ослаблением активации транскрипции генов-мишеней
- ТВХ5 играет ключевую роль в инициации роста верхних конечностей
- Генетические факторы
о Аутосомно-доминантный тип наследования:
- Риск появления заболевания у потомства носителей составляет 50%
о Практически полная пенетрантность
о Вариабельная экспрессивность
о Генотипирование ТВХ5 обладает высокими показателями чувствительности и специфичности в отношении синдрома Холт-Орама только при строгом соблюдении диагностических критериев:
- Мутации гена ТВХ5 обнаруживаются по меньшей мере у 70% пациентов с синдромом Холт-Орама и преаксиальными дефектами радиального луча
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о Дефекты радиального луча различной степени тяжести
о Порок сердца
• Другие субъективные и объективные симптомы в постнатальном периоде:
о ДМПП, дефект вторичной перегородки (ostium secundum)
о Нарушения ритма и проводимости сердца
2. Демографические особенности:
• 1:100 000 детей, рожденных живыми
• 85% случаев - мутации de novo
3. Естественное течение и прогноз:
• Общий прогноз зависит от тяжести порока сердца и степени поражения верхних конечностей
• Возможна нормальная продолжительность жизни
• Отставание в умственном развитии наблюдают не чаще, чем в среднем в популяции
4. Лечение синдрома Холт-Орама:
• В пренатальном периоде:
о Генетическое консультирование
о При наличии у родителей установленной мутации ТВХ5 возможна пренатальная диагностика с помощью амниоцентеза или биопсии ворсин хориона
о Преимплантационная генетическая диагностика возможна только при наличии установленной мутации
• В постнатальном периоде:
о Междисциплинарный подход к ведению заболевания
о Хирургическое лечение пороков сердца
о Ортопедическое лечение поражений кисти, направленное на улучшение ее функции:
- Поллицизация II пальца кисти или стопы для воссоздания утраченного пальца кисти
о Наблюдение:
- Электрокардиография (1 раз в год)
- При наличии нарушений проводимости сердца - холте-ровское мониторирование (1 раз в год)
- ЭхоКГ (1 раз в 1-5 лет)
е) Список использованной литературы:
1. Al-Qattan ММ et al: Molecular basis of the clinical features of Holt-Oram syndrome resulting from missense and extended protein mutations of the TBX5 gene as well as TBX5 intragenic duplications. Gene. 560(2): 129-36, 2015
2. Wall LB et al: Defining features of the upper extremity in Holt-Oram syndrome. J Hand Surg Am. 40(9):1764-8, 2015
3. Barisic I et al: Holt Oram syndrome: a registry-based study in Europe. Orphanet J Rare Dis. 9:156, 2014
Читайте также:
- Применение интерферона. Побочные эффекты интерферонов
- Лучевая диагностика несеминомной герминативноклеточной опухоли средостения на рентгене, КТ
- Гомеостатические механизмы при гиперосмотической гипергидратации. Синдром Кона. Гиперосмотическая гипергидратация.
- Диагностика церебральной малярии по КТ, МРТ
- Психиатрия - симптомы и лечение