УЗИ при синдроме Беквита-Видемана у плода
Обновлено: 18.05.2024
Кафедра акушерства, гинекологии, перинатологии и репродуктологии факультета последипломного профессионального обучения врачей Московской медицинской академии им. И.М. Сеченова;
Научный центр акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова
Центр лечения бесплодия "ЭКО", Москва
Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай)
Журнал: Проблемы репродукции. 2014;(3): 58‑61
Назаренко Т.А., Зыряева Н.А. Рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ (клинический случай). Проблемы репродукции. 2014;(3):58‑61.
Nazarenko TA, Zyriaeva NA. The birth of the child with Beckwith-Wiedemann syndrome after IVF/ICSI (a case report). Russian Journal of Human Reproduction. 2014;(3):58‑61. (In Russ.).
Представлено клиническое наблюдение - рождение ребенка с синдромом Видемана-Беквита у пациентки после применения программы ЭКО/ИКСИ.
В основе импринтинга лежат различия в экспрессии генов, обусловленные метилированием ДНК. У человека предполагают наличие 300-500 импринтированных генов. В основном они кодируют ростовые факторы, одни экспрессируют с отцовской, другие - с материнской хромосомой 6. Клетки каждого человека содержат отпечатки (характер метилирования) от обоих родителей. При гаметогенезе эти отпечатки стираются и устанавливается характер метилирования, присущий женскому или мужскому организму. При оплодотворении эмбрион получает по одному набору от каждого родителя с соответствующим характером экспрессии. В случае попадания в процессе оплодотворения двух гомологичных хромосом от одного родителя это состояние называется однородительской дисомией (ОРД) [5, 6].
Нарушения метилирования возможны при гаметогенезе, в процессе оплодотворения и преимплантационного развития. Нарушения метилирования могут происходить на разных уровнях организации генома (геномный, хромосомный, генный), вызывая соответствующие отклонения в развитии или болезни импринтинга [4, 6, 7]. Наиболее распространены и изучены у человека синдромы Прадера-Вили, Ангельмана, Видемана-Беквита, Сильвера-Рассела и некоторые другие.
Синдром Видемана-Беквита, или EMG-синдром по первым буквам характерной триады - омфалоцеле, макроглоссия, гигантизм, впервые описан патологом J. Beckwith (США) в 1963 г. и педиатром H.-R. Wiedemann (Германия) в 1964 г. Синдром встречается с частотой 1 случай на 10-12 тыс. новорожденных, при этом 85% - спорадические случаи и только 15% - наследственные (аутосомно-доминантный тип наследования с неполной пенетрантностью, передача по материнской линии) [8].
Критерии диагностики в настоящее время четко не установлены, но диагноз ставится при наличии 3 больших и 1 малого диагностического критерия. Из больших критериев наиболее характерны макросомия, макроглоссия, омфалоцеле, аномалии почек, эмбриональные опухоли. Из малых критериев характерны многоводие, неонатальная гипогликемия. Дифференциальный диагноз проводится со следующими синдромами: синдромом Симпсона-Голаби-Бехмеля (макросомия, висцеромегалия, макроглоссия, аномалии почек; Х-сцепленный рецессивный тип наследования), синдромом Перлмана, синдромом Костелло, синдромом Сотоса, синдромом Марото-Лами (мукополисахаридоз IV типа) [8].
Молекулярной основой синдрома Видемана-Беквита являются нарушения в генах короткого плеча хромосомы 11. Участок на коротком плече 11-й хромосомы включает две группы импринтированных генов - домен 1 (гены ИПФР2, Н19 и ЦИ1) и домен 2 (гены CDKN1C, KCNQ1 и ЦИ2) (ЦИ - центр импринтинга) [8].
Этиология синдрома сложна и включает 5 групп нарушений. Нарушения метилирования 1 и 2 ЦИ (потеря метилирования ЦИ2 материнского аллеля CDKN1C - 50% и гиперметилирование ЦИ1 материнского аллеля ИПФР2 и Н19 - 5%), а также отцовская ОРД по 11-й хромосоме - 20% - встречаются наиболее часто. Нередки мутации гена CDKN1C материнского аллеля: спорадические - 5%, наследственные - 40%. Также встречаются дупликации, инверсии, транслокации в этих доменах - 1% и субмикроскопические нарушения - частота не определена. В 20% случаев характер нарушений не известен [8].
Учитывая возможность нарушений метилирования в процессе гаметогенеза, оплодотворения и преимплантационного развития, встает закономерный вопрос о влиянии вспомогательных репродуктивных технологий (ВРТ) на частоту возникновения болезней импринтинга. По данным литературы 12, многие авторы отмечают повышенный риск болезней импринтинга после применения ВРТ. Некоторые авторы [15, 16] не отмечают повышения риска. Другие связывают болезни импринтинга у потомства с исходными нарушениями у родителей при бесплодии [17]. Также имеются отдельные клинические наблюдения [18, 19].
Представляем клиническое наблюдение.
Пациентка И., 1980 года рождения, рост 176 см, масса 62 кг. Наследственность не отягощена. Из соматического анамнеза обращает на себя внимание наличие пролапса митрального клапана, гипотонии и с 12 лет брадикардии. В детстве состояла на учете по поводу хронического пиелонефрита, с учета снята: аппендэктомия в 16 лет; страдает хроническим колитом. Является носителем вируса простого герпеса (ВПГ1), периодически отмечает обострения лабиального герпеса. Менархе в 12 лет, менструальный цикл регулярный. Половая жизнь с 18 лет. Беременностей не было. Брак первый с 2008 г., муж - 1978 года рождения, здоров. Гинекологический анамнез отягощен оперативными вмешательствами: 2 раза было экстренное чревосечение по поводу разрыва эндометриоидных кист яичников (в 2004 и 2008 г.). В 2006 г. была проведена лапароскопия, разделение спаек в малом тазу и гистерорезектоскопия, рассечение неполной внутриматочной перегородки. В 2012 г. - гистероскопия, раздельное диагностическое выскабливание перед проведением повторного цикла ЭКО.
Женщина обратилась по поводу бесплодия в 2008 г. Диагноз: первичное бесплодие; наружный генитальный эндометриоз IV степени; спаечный процесс в малом тазу; седловидная матка; хронический эндометрит.
Проведено 3 цикла ЭКО/ИКСИ по длинному протоколу (агонисты ГнРГ + чМГ + рФСГ), при пункциях получено по 3 ооцита, проведен перенос 2 эмбрионов на 3-и сутки, беременность наступила в 3-м цикле, одноплодная.
Беременность протекала с угрозой прерывания в I триместре, с 23 нед выявлено многоводие, с 27 нед - двусторонний уретерогидронефроз у плода по данным УЗИ. В 34 нед беременности отмечено многоводие крайней степени выраженности по данным УЗИ, нарушение кровотока у плода по данным допплеровского исследования, произведено кесарево сечение в экстренном порядке, родился живой недоношенный мальчик массой тела 2850 г, длиной 46 см, оценка по шкале Апгар 3-7 баллов. Мама выписана на 10-е сутки, послеоперационый период протекал без осложнений.
Мальчик родился в октябре 2009 г. Диагноз основной: множественные врожденные пороки развития; порок центральной нервной системы (незрелость структур головного мозга); макросомия; макроглоссия; неонатальный сахарный диабет; правосторонний пузырно-мочеточниковый рефлюкс III-IV степени; дефект межжелудочковой перегородки. Осложнения: церебральная ишемия II степени; геморрагический синдром (легочное и желудочное кровотечение). Сопутствующий: тяжелая асфиксия при рождении; внутриутробная пневмония; анемия новорожденных; недоношенность 34 нед.
В родильном зале ребенку произведена интубация трахеи, переведен на искусственную вентиляцию легких. Ребенок наблюдался в течение 1 мес жизни в отделении интенсивной терапии новорожденных. Отмечались плотные распространенные отеки туловища, синдром угнетения ЦНС (снижен мышечный тонус и угнетены рефлексы), в последующем на фоне терапии отмечена положительная динамика. По поводу гипергликемии с рождения получал инсулинотерапию (актропид внутривенно капельно), был взят генетический анализ на неонатальный сахарный диабет. Был установлен уретральный катетер, затем наложена пункционная цистостома, после чего признаки дилатации чашечно-лоханочной системы и обоих мочеточников по данным УЗИ купированы.
По данным магнитно-резонансной томографии (МРТ) выявлено нарушение формирования структур головного мозга: картина наружной гидроцефалии, гипоплазии височных долей, нарушения процесса формирования борозд головного мозга, задержки миелинизации. При консультации генетиком на основании наличия синдромальной формы макросомии и множественных врожденных пороков развития поставлен диагноз: синдром Симпсона-Голаби-Бехмеля. Взят цитогенетический анализ. Кариотип 46XY, нормальный мужской.
В возрасте 1 мес ребенок переведен в ДГКБ №1. На первом году жизни произведены операции по поводу паховой грыжи, глоссомегалии. В 7 мес жизни на консультации генетика установлен диагноз: синдром Видемана-Беквита (макросомия, макроглоссия, задержка психомоторного развития - гипоплазия височных долей по данным МРТ). Ребенок наблюдался у невролога, челюстно-лицевого хирурга. В 2 года на консультации генетика поставлен диагноз: синдром Видемана-Беквита (макроглоссия, мегауретер с гидронефротической трансформацией. Аномалия головного мозга на МР-томограмме. Задержка темпов психомоторного развития. Гипертензионно-гидроцефальный синдром). Ребенок получал лечение у нефролога, невролога, проходил скрининг 4 раза в год по поводу возможных эмбриональных опухолей (УЗИ, альфа-фетопротеин крови).
Пациентка повторно обратилась с целью достижения беременности в августе 2012 г. Естественно возник вопрос о возможностях профилактики и диагностики синдрома Видемана-Беквита при повторной беременности у женщины. Пациентка была проконсультирована генетиками из Медико-генетического центра РАМН. К сожалению, в настоящее время точная лабораторная диагностика болезней импринтинга четко не разработана, поэтому молекулярно-генетическое обследование ребенка и супружеской пары не проводилось. При медико-генетическом консультировании синдром Видемана-Беквита у ребенка был расценен как спорадический, поэтому риск при повторной беременности был расценен как низкий.
Учитывая упомянутые молекулярные особенности, лежащие в основе болезней импринтинга, проведение преимплантационной генетической диагностики (ПГД) также не представлялось возможным.
Проведен 4-й цикл ЭКО - по длинному протоколу (агонисты ГнРГ, чМГ, рФСГ), при трансвагинальной пункции получено 2 ооцита, 27.09.12 перенесено 2 эмбриона 3-го дня развития, наступила вторая беременность (последняя менструация 09.09.12).
В I триместре у женщины был впервые выявлен первичный субклинический гипотиреоз, в течение всей беременности пациентка принимала эутирокс 75-50 мкг/сут. В остальном беременность протекала без особенностей.
По результатам 1-го и 2-го скринингов и УЗИ отклонений от нормы не отмечено. При медико-генетическом консультировании показаний для проведения инвазивной пренатальной диагностики не выявлено. Кроме того, в отношении болезней импринтинга возможности пренатальной диагностики также четко не установлены.
Согласно информации, полученной из источников в Интернете, методы лабораторной диагностики синдрома Видемана-Беквита, имеющиеся на современном этапе развития медицины, находятся на стадии разработки и лишь частично внедрены в практику, в основном в США, странах Европы. Методы лабораторной диагностики зависят от генетической этиологии и включают следующие способы: 1) анализ метилирования с помощью метилчувствительной множественной полимеразной цепной реакции (МЧ мПЦР; MS-MLPA); МЧ ПЦР; 2) анализ однородительской дисомии с помощью исследования однонуклеотидного полиморфизма, МЧ мПЦР (MS-MLPA) и ПЦР; 3) определение мутаций с помощью анализа последовательностей; 4) определение дупликаций, инверсий и транслокаций с помощью цитогенетического анализа и FISH и, возможно, современного метода сравнительной геномной гибридизации; 5) выявление субмикроскопических нарушений с помощью анализа микроделеций и микродупликаций [7, 8, 20, 21].
Тактика медико-генетического консультирования также зависит от генетической причины синдрома Видемана-Беквита, последняя определяет и степень риска для сибсов пробанда [8].
У 85% больных с синдромом Видемана-Беквита наследственность не отягощена и кариотип нормальный. При этом высокий риск (50%) существует при наличии мутаций гена CDKN1C и микроделеций, микродупликаций; в остальных случаях риск низкий. В этих случаях проводят определение мутаций у пробанда и родителей, а также в семье.
При невыясненной причине (мозаицизм при ОРД) риск эмпирически низкий.
У 10-15% больных наследственность отягощена и нормальный кариотип.
При выявлении мутации CDKN1C у пробанда (40%): при наличии мутации у одного из родителей риск составляет 50%, при отсутствии мутации у родителей риск низкий. Но возможен мозаицизм. При отсутствии мутации CDKN1C у пробанда (60%) риск для сибсов составляет до 50%.
Пренатальная диагностика проводится следующим образом [8].
В случае наличия больного ребенка в семье: при выявлении мутаций проводят амниоцентез и анализ ДНК. Теоретически возможен анализ метилирования в амниоцитах. При всех беременностях риска: в 16 нед при наличии омфалоцеле определяют альфа-фетопротеин крови, проводят УЗИ в 19-20 нед и 25-32 нед беременности для выявления пороков развития и макросомии.
При неотягощенной наследственности: в случае выявления изолированного омфалоцеле при УЗИ возможен амниоцентез, анализ метилирования и мутации гена СDKN1С, дупликации, инверсии, транслокации. Проводят динамическое УЗИ во время беременности для выявления пороков развития и макросомии.
У новорожденного проводят мониторинг неонатальной гипогликемии. Диагноз подтверждают вышеуказанными способами [8].
Вопрос о преимплантационной диагностике не решен. Теоретически возможно применение метода сравнительной геномной гибридизации в случае отягощенной наследственности и наличия мутаций в семье.
Омфалоцеле
Омфалоцеле - врожденная аномалия передней брюшной стенки, при которой органы брюшной полости выходят за ее пределы в составе грыжевого мешка. Клинически патология проявляется эвентрацией петель кишечника, желудка, печени и других органов, прикрытых висцеральной брюшиной, через грыжевые ворота в участке пупочного кольца. Антенатальная диагностика включает в себя УЗИ ОБП, определение уровня α-фетопротеина, амниоцентез с дальнейшим кариотипированием. Лечение хирургическое, предусматривает радикальное или поэтапное погружение содержимого грыжевого пешка обратно в брюшную полость с последующей пластикой передней брюшной стенки.
Общие сведения
Омфалоцеле (эмбриональная грыжа, грыжа пупочного канатика) - аномалия развития, при которой происходит выпячивание органов брюшной полости, укрытых висцеральной брюшиной, сквозь переднюю брюшную стенку. Впервые данную патологию описал французский хирург А. Паре в 1634 году. В среднем заболеваемость составляет порядка 2,2:10000 новорожденных. Точную частоту, с которой встречается омфалоцеле, установить невозможно, поскольку в большинстве случаев такие беременности прерываются. У мальчиков данная патология возникает в 1,5 раза чаще, чем у девочек. Наибольшая склонность наблюдается у представителей европеоидной расы - 85% от всех случаев омфалоцеле. Для представителей негроидной расы этот показатель составляет 13%, монголоидной - 2%. Примерно 55% случаев патологии диагностируется при беременности у женщин в возрасте старше 35 лет. Общая летальность зависит от сопутствующих расстройств и колеблется от 9 до 60% новорожденных. При изолированной форме и адекватном лечении прогноз для жизни и здоровья ребенка благоприятный.
Причины омфалоцеле
Омфалоцеле - это гетерогенное заболевание, при котором нарушается процесс внутриутробного вправления физиологической пупочной грыжи. Патогенетически это может быть результатом пороков развития пупочного кольца и передней брюшной стенки, генетических аномалий, неполного погружения органов обратно в брюшную полость или дефектов строения кишечника.
При внутриутробном развитии кишечника плода, а именно - в процессе его трансформации от первичной кишечной трубки до зрелых петель происходит его физиологической разворот. Данный процесс начинается с 5 недели беременности. От этого момента и до 10 недели кишечник сильно увеличивается в объеме, из-за чего не помешается в брюшной полости. Петли давят на брюшную стенку, формируя физиологическую пупочную грыжу. К концу 10 недели абдоминальная полость стремительно прибавляет в объеме, на фоне чего происходит самостоятельное вправление эвентрации. Если данный механизм нарушается силу различных патологических изменений, возникает омфалоцеле.
Способствовать возникновению данной патологии могут вредные привычки матери (алкоголь, курение, наркотики), нерациональный прием медикаментов, беременность после 35 лет, при которой растет риск хромосомных аномалий - синдромов Патау, Эдвардса. Довольно часто омфалоцеле выступает в роли одного из симптомов таких патологий, как синдром Беквита-Видемана, пентада Кантрелла, синдром амниотических тяжей, порок развития стебля тела, OEIS комплекса. Крайне редко удается установить наследственную склонность, что свидетельствует о возможной генетической предрасположенности.
Классификация и симптомы омфалоцеле
В педиатрии в зависимости от размера дефекта и его содержания выделяют следующие формы омфалоцеле:
- Малая. Диаметр - до 5 см. Наиболее распространенная форма. Содержит 1-2 кишечные петли. В основном выступает в роли проявлений хромосомных аномалий.
- Средняя. Размер дефекта пупочного кольца - от 5 до 10 см. В составе грыжевого мешка содержатся 2-4 кишечные петли.
- Большая. Дефект передней брюшной стенки составляет более 10 см. Помимо петель кишечника через него выходит часть печени, желудок и другие органы.
По наличию сопутствующих патологий выделяют:
- Изолированное омфалоцеле. Грыжа пупочного канатика - единственная развившаяся внутриутробная патология.
- Сочетанная форма. Помимо дефекта пупочного кольца у ребенка присутствуют хромосомные мутации (25-35%), пороки развития сердечно-сосудистой (15-50%) и мочеполовой систем (до 15%). Характерны грыжи пищеводного отверстия диафрагмы и дисплазии тазобедренных суставов, другие скелетные аномалии.
Омфалоцеле является врожденной патологией, которая диагностируется еще в антенатальном периоде. После родов общее состояние ребенка зависит от сопутствующих заболеваний и срока гестации. При омфалоцеле характерны преждевременные роды, масса тела ребенка 1500г и меньше. Объективные признаки визуализируются уже с момента рождения. Определяется дефект передней брюшной стенки, который локализирован по срединной линии на уровне пупочного кольца. В этом месте находится образование, представленное петлями тонкого и толстого кишечника, возможно - желудком и печенью, покрытыми висцеральными листком брюшины. В 10-20% наблюдается анте- или интранатальный разрыв грыжевого мешка. Пуповина также входит в состав омфалоцеле. Абдоминальные мышцы развиты нормально. Другие возможные пороки развития зависят от сопутствующих патологий и могут включать в себя деформации позвоночного столба и конечностей, макроглоссию, макросомию, атрезию ануса и т. д.
Диагностика омфалоцеле
В современных условиях наличие омфалоцеле определяется еще в I триместре беременности благодаря инструментальным и лабораторным методам исследования. Ведущую роль играет УЗ-диагностика. В зависимости от аппарата установить наличие данной патологии можно уже на 11-14 неделе. Основной признак - эвентрация петель кишечника, печени, желудка за пределы брюшной полости в участке крепления пуповины к брюшной стенке. Все вышедшие органы покрыты мембраной, состоящей из брюшины, вартонового студня и амниотической оболочки. Наличие данной мембраны важно для проведения дифференциальной диагностики с гастрошизисом. При внутриутробном разрыве грыжевого мешка петли кишечника прикрывает только амнион. Также на потенциальное развитие омфалоцеле может указывать размер ворот физиологической грыжи, составляющий свыше 7 мм в диаметре. УЗИ позволяет выявить другие проявления присутствующих генетических патологий. Повторные исследования проводятся каждые 2-3 недели, т. к. возможно самостоятельно закрытие дефекта в более поздние сроки.
Лабораторная диагностика омфалоцеле осуществляется при помощи биохимического исследования крови с измерением уровня α-фетопротеина. При врожденных пороках развития этот показатель будет значительно выше нормы для имеющегося срока беременности. Для подтверждения генетических аномалий показано проведение амниоцентеза с дальнейшим кариотипированием.
Лечение омфалоцеле
При антенатальной постановке диагноза омфалоцеле роды проводятся в специализированном перинатальном центре в условиях развернутой операционной. При малых и средних размерах дефекта пупочного кольца и отсутствии угрожающих жизни матери и ребенка состояний родоразрешение может осуществляться через естественные родовые пути. Во всех других ситуациях показано кесарево сечение в связи с большим риском интранатального разрыва грыжевого мешка. Дальнейшая терапевтическая тактика зависит общего состояния ребенка, размеров пупочной грыжи и сопутствующих патологий.
Консервативная терапия используется при невозможности провести хирургическое вмешательство. Как правило, она применяется при больших формах омфалоцеле и комбинации с множественными тяжелыми аномалиями развития. Лечение заключается в формировании плотной корки и рубца, что трансформирует данную патологию в массивную вентральную грыжу, которую в дальнейшем оперируют. С этой целью применяют дубящие средства (5% перманганат калия, нитрат серебра), которые наносят на вертикально зафиксированный за пуповину грыжевой мешок 2-3 раза в день. Консервативное лечение используется крайне редко, т. к. при нем имеется большая вероятность инфицирования и сепсиса, риск разрыва оболочек, массивной спаечной болезни в дальнейшем. Предпочтение отдается раннему оперативному вмешательству.
Хирургическая тактика при омфалоцеле напрямую зависит от размеров дефекта брюшной стенки, операции могут проводиться в один или несколько этапов. Начинают такое лечение на протяжении первых 1-2 дней жизни ребенка. При одномоментном вмешательстве выполняется погружение содержимого грыжевого мешка в абдоминальную полость и послойное зашивание брюшной стенки с ее пластикой и формированием пупка. Такая тактика - метод выбора, при омфалоцеле малых и средних размеров с минимальной торакоабдоминальной диспропорцией. В других случаях прибегают к поэтапному лечению. Первый этап - подшивание силиконового мешка с силопластиковым покрытием и помещением в него содержимого грыжевого мешка. По мере постепенного погружения органов в абдоминальную полость данный мешок перевязывают, уменьшая его в объеме. На 5-15 день выполняется второй этап - удаление мешка и формирование минимальной вентральной грыжи посредством ушивания дефекта. В возрасте 5-7 месяцев эта грыжа удаляется, после чего осуществляется полноценная пластика брюшной стенки.
После первого этапа или одноэтапной операции ребенок помещается в кувез для поддержания температурного режима, применяются обезболивающие, антибиотики широкого спектра действия. Режим питания парентеральный. При необходимости выполняется декомпрессия желудка через назо- или орогастральный зонд, проводится инфузионная терапия и ИВЛ. После проведенного лечения следует длительный реабилитационный период, в течение которого осуществляется коррекция всех послеоперационных осложнений.
Прогноз и профилактика омфалоцеле
В целом прогноз для детей с омфалоцеле сомнительный. Исход зависит от срока гестации, сопутствующих хромосомных аномалий и анатомических пороков развития, диаметра дефекта пупочного кольца, объема и содержания грыжевого мешка, результатов проведенного лечения. Как правило, при адекватной терапии и отсутствии сопутствующих патологий, несовместимых с жизнью, вероятность благоприятного исхода достаточно велика. При изолированной форме омфалоцеле малого размера, несмотря возможные отдаленные осложнения хирургического лечения (ГЭРБ, спаечная болезнь, пупочная грыжа), дальнейший рост и развитие ребенка не будут отличаться от возрастной нормы. На протяжении всего периода реабилитации показано диспансерное наблюдение у лечащего педиатра, хирурга и семейного врача.
Профилактика заключается в медико-генетическом консультировании семейных пар, планировании беременности, полном отказе матери от вредных привычек. С целью ранней диагностики омфалоцеле необходимо регулярно посещать женскую консультацию и проходить соответствующие обследования: УЗИ и измерение α-фетопротеина крови.
Проблемы с пуповиной плода - УЗИ с петлей на шее

Патологии пуповины (пупочного канатика), соединяющей ребенка с организмом матери, по статистике наблюдаются более, чем у половины беременных. Их коварство заключается в отсутствии симптомов до начала родовой деятельности и высоком риске гибели новорожденного во время родов.

Для чего нужна пуповина
Пуповина служит для снабжения ребенка кислородом и питательными веществами. Для этого в канатике есть две артерии и вены. Природа позаботилась о защите пуповины от повреждающих факторов. Снаружи орган покрыт плодными оболочками, а внутри содержит вартонов студень, гасящий толчки и случайные удары в область живота.
Орган формируется на 2-3-й неделе и увеличивается по мере роста плода. К родам длина канатика практически равна росту малыша, составляя 50-60 см. Диаметр трубки тоже растет, постепенно увеличиваясь до 2 см.
Прикрепляется пуповина к плаценте (детскому месту) в центре или сбоку. Иногда наблюдается прикрепление к оболочкам (оболочечное). Такая ситуация увеличивает риск возникновения плацентарной недостаточности.
Почему возникают проблемы с пуповиной
Этот вопрос до сих пор еще досконально не изучен, но предполагается, что часть патологий закладывается при формировании плодного яйца и имеет генетическую причину.
Частая причина возникновения узлов и обвития пуповиной — многоводие. В большом объеме амниотической жидкости движения плода не ограничены и он намного легче проскакивает в пуповинную петлю. Провоцируют ситуацию гипоксия плода и повышенный тонус матки, заставляющие ребенка двигаться интенсивнее, а также слишком длинная пуповина.
Поэтому обнаружение на УЗИ плода двух и даже трёхкратного обвития, когда ребёнок находится внутри мощной пуповинной петли, часто указывает на гипоксию, требующую дополнительных обследований. Женщине назначается Кардиотокография (КТГ), показывающая состояние плода путём регистрации частоты его сердечных сокращений.
В норме ЧСС составляет:
- 9 -10 недель. — 170-190 уд/мин;
- с 11 недель - 140-160 уд/мин.
Угнетение сердечной деятельности указывает на гипоксию. Если не принять мер, малыш потеряет силы и перестанет шевелиться. Возникнет выраженная нехватка кислорода, приводящая к нарушению внутриутробного развития и даже мертворождению.
Аномалии развития пуповины
Отсутствие второй пуповинной артерии . Эта патология образуется еще на этапе формирования плодного яйца. Такую беременность можно доносить, но зачастую аномалия сочетается с врожденными патологиями развития плода.
В этом случае беременная женщина нуждается в дополнительном УЗИ -контроле, позволяющем определить возможные генетические аномалии. Для получения более информативных данных о развитии ребенка рекомендуется проведение 3D и 4D-скрининга. При обнаружении тяжелых пороков рекомендуется прерывание беременности на ранних сроках.
Укорочение пуповины не влияет на развитие малыша, но создает проблемы в родах, заканчиваясь отслоением плаценты или гипоксией из-за натяжения вен пупочного канатика. Иногда малыш просто не может выйти наружу. Роды могут закончиться разрывом сосудов и массивным кровотечением.
Определить длину пупочного канатика можно только на последнем скрининге, проводимом перед родами. При обнаружении такой аномалии назначается родоразрешение путем кесарева сечения.
Чрезмерно длинная пуповина опасна частым возникновением обвития и пуповинного предлежания, когда ее петля находится на выходе из половых путей перед головкой ребенка. Еще одно осложнение - выпадение пуповинной петли в родах. При возникновении угрозы жизни мамы и малыша проводятся экстренные оперативные роды.
Обвития и узлы
В этом случае пуповина развита нормально, но обвита вокруг плода или завязана узлом. Ситуация, как уже обозначалось, возникает, когда чрезмерно активный плод проскакивает в созданную им же петлю. Количество «витков» бывает разным и зависит от дины пупочного канатика. В науке описан случай девятикратного обвития.
Ситуация грозит затягиванием пуповинных петель на шее малыша в родах, приводящему к родовым травмам и даже к смерти. Длины такой пуповины не хватит, и она не даст плоду родиться. Единственный выход - оперативное родоразрешение. Самостоятельные роды разрешаются, если обвитие однократное и не тугое.
Пуповиный узел - опасная патология, ведь малыш, двигаясь в утробе мамы, может затянуть его, окончательно лишив себя кислорода и питательных веществ. В этом случае женщине понадобиться постоянное наблюдение, возможно даже в условиях стационара.
От истинных узлов нужно отличать ложные, когда пупочный канатик утолщен за счет скопления вартонова студня или патологического расширения сосудов. В этом случае здоровью ребенка ничего не угрожает. Определить тип узла можно на качественном УЗИ аппарате, дающем детальную картинку.
Неправильное прикрепление пуповины
В этом случае пуповинный канатик крепится не к плаценте, а к плодным оболочкам, что усиливает риск его отрыва во время родов. При подозрении на патологию женщине проводится цветной допплер (ЦДК), выявляющий патологию практически в 100% случаев.
Особенно опасно, если патология сочетается с укорочением пуповины или обвитием. В этом случае показано оперативное родоразрешение. Но самый тяжелый случай неправильного прикрепления пуповинного канатика - омфалоцеле. При этой патологии кишечник и внутренние органы ребенка выходят за пределы его брюшной стенки, образуя грыжевой мешок, к которому прикреплена пуповина. Риск гибели малыша очень велик.
Омфалоцеле — частый признак тяжелых врожденных патологий - синдромов Беквита-Видемана, Эдвардса Патау, пентады Кантрелла. У половины таких детей наблюдаются пороки развития суставов, сердца, внутренних органов. Поэтому при обнаружении отклонения нужно провести тщательный скрининг на хромосомные патологии. При подтверждении тяжелых диагнозов женщине предлагается прерывание беременности.
Если ребенок в остальном здоров, проводится оперативное родоразрешение в срок. В дальнейшем малышу понадобится несколько операций по вправлению органов и удалению грыжевого мешка.
Обнаружение проблем с пуповиной дает возможность выбрать нужную тактику ведения беременности и родов, позволяющую избежать тяжелых осложнений, сохранив жизнь и здоровье маме и малышу.
УЗИ при синдроме Беквита-Видемана у плода
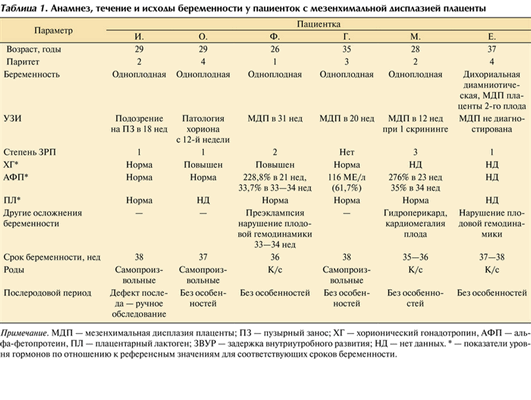
Возраст женщин с одноплодной беременностью составил 26—35 лет (табл. 1), Таблица 1. Анамнез, течение и исходы беременности у пациенток с мезенхимальной дисплазией плаценты Примечание. МДП — мезенхимальная дисплазия плаценты; ПЗ — пузырный занос; ХГ — хорионический гонадотропин, АФП — альфа-фетопротеин, ПЛ — плацентарный лактоген; ЗВУР — задержка внутриутробного развития; НД — нет данных. * — показатели уровня гормонов по отношению к референсным значениям для соответствующих сроков беременности. во всех случаях беременность наступила спонтанно. Одна женщина была первородящей, остальные имели в анамнезе срочные самопроизвольные роды (масса новорожденных 3180—4040 г, все дети здоровы).
Пациентка 37 лет с двойней имела в анамнезе срочные оперативные роды и 2 внематочные беременности; настоящая беременность наступила в результате экстракорпорального оплодотворения и переноса эмбриона.
Течение настоящей беременности у первородящей пациентки осложнилось преэклампсией умеренной степени, еще у одной пациентки отмечены кардиомегалия и гидроперикард плода. Задержка внутриутробного развития разной степени отмечена в 5 наблюдениях.

Изменение плаценты было впервые диагностировано по результатам УЗИ на разных сроках беременности — 12—31 нед. В большинстве случаев был первично поставлен диагноз ЧПЗ или подозрение на него. При обследовании в МОНИИАГ по результатам УЗИ диагностирована МДП (рис. 1, а, Рис. 1. Мезенхимальная дисплазия плаценты. а, б — ультразвуковые признаки: а — диффузное утолщение и множественные кистозные включения в плаценте. Структура плаценты изменена, имеется множество жидкостных включений различного диаметра от 4 до 12 мм. Включения в основном овоидной и округлой формы, границы ровные, четкие. Граница плацентарной ткани и миометрия отчетливая; б — в структуре плацентарной ткани множественные локусы кровотока. Плацентарные сосуды большого диаметра. Архитектоника сосудов плаценты не изменена; в, г — макроскопические изменения плацент: крупные сосуды хориальной пластинки и стволовых ворсин, кисты разных размеров. б).
Уровень чХГ был нормальным у 2 пациенток, повышенным — у 2. К сожалению, показатели еще 2 пациенток не известны. Уровень АФП у 3 пациенток был нормальным, у 2 имел сходную динамику: высокий уровень белка (до 275%) на сроке 21—23 нед со снижением до 33—35% к сроку 33—34 нед (см. табл. 1).
Роды самопроизвольные в 3 случаях, в 3 произведено кесарево сечение, показаниями к которому явились: нарастание тяжести преэклампсии, нарушение гемодинамики плода, несостоятельность рубца после кесарева сечения (пациентка с двойней). Срок родоразрешения составил 35—38 нед. У 1 пациентки проведено ручное обследование в связи с дефектом последа, других осложнений послеродового периода у пациенток не отмечено.

Лишь 1 ребенок родился с нормальной массой (3580 г при сроке беременности 38 нед), в остальных случаях отмечена задержка роста плода 1—3-й степени (табл. 2). Таблица 2. Состояние новорожденных и макроскопическая характеристика плацент при мезенхимальной дисплазии Примечание. ЗВУР — задержка внутриутробного развития; РДС — респираторный дистресс-синдром; ДМПП — дефект межпредсердной перегородки; ППК — плацентарно-плодовый коэффициент; НД — нет данных. * — масса девочки 1-й из двойни 2820 г, длина 47 см. Оценка по шкале Апгар у 4 детей составила 8—9 баллов, у 2 — 7—8 баллов. Соотношение полов м/ж составило 3:3. Ранний неонатальный период протекал без осложнений у 3 детей, у 3 было диагностировано перинатальное поражение ЦНС 1-й степени, у одного из них в сочетании с респираторным дистресс-синдромом, у второго — с респираторным дистресс-синдромом и дефектом межпредсердной перегородки.
Масса плацент при МДП была существенно выше средней от 771 до 1498 г, что составило 95,8—100‰ (см. табл. 2). Лишь в 1 случае, у пациентки с преэклампсией, масса плаценты соответствовала средним показателям, составляя 51‰. Выраженные различия выявлены между плацентами новорожденных из двойни (масса одного плода 2820 г, плаценты 489 г, масса второго плода 2560 г, плаценты 771 г при сроке беременности 37—38 нед). В 3 случаях имелось краевое или оболочечное прикрепление пуповины. Несмотря на большую массу плацентарного диска, во всех наблюдениях толщина пуповины не превышала 1,5 см, а в половине случаев была менее 1 см. Хориальная пластинка с выбухающими резко извитыми сосудами с широким просветом, в паренхиме определялись кистозные полости со светлым прозрачным содержимым, белесоватые тяжи с крупными сосудами в них. Степень изменений значительно варьировала (см. рис. 1, в, г).
При микроскопическом исследовании основные изменения наблюдались в стволовых ворсинах: резкое увеличение их размеров, отек стромы разной степени выраженности во многих случаях с формированием кистозных полостей (рис. 2, а, б). Сосуды в пораженных ворсинах присутствовали, но их количество и размеры значительно варьировали (см. рис. 2, в, г). Периферические отделы ворсинчатого дерева были сформированы правильно, представлены дифференцированными промежуточными и терминальными ворсинами в разных соотношениях (см. рис. 2, д).

Рис. 2. Микроскопические изменения плацент при мезенхимальной дисплазии. а, б — отечные кистозно-измененные стволовые ворсины с сохранными сосудами, без пролиферации трофобласта; в, г — разная степень васкуляризации и отека гигантских ворсин; д — нормально сформированные терминальные ворсины рядом с кистозно-измененной стволовой ворсиной (указана стрелкой); е — плацента пациентки с преэклампсией: в левом нижнем углу фрагмент гигантской стволовой ворсины, терминальные ветви мелкие, хаотично ветвящиеся, с узкими немногочисленными сосудами. Окраска гематоксилином и эозином. а, г — ×50, в, е — ×100, б, д — ×200.

Рис. 2. Микроскопические изменения плацент при мезенхимальной дисплазии. ж, з— варианты патологического строения сосудов. Иммуногистохимическая реакция с антителами к СD34; и — иммуногистохимическая реакция с антителами Ki-67. Позитивная ядерная реакция в единичных клетках цитотрофобласта и стромы ворсин; к — скопление фибрина в межворсинчатом пространстве. Окраска гематоксилином и эозином; л — пристеночный тромб в сосуде хориальной пластинки. Окраска MSB; м — фетальная тромботическая васкулопатия, группа бессосудистых ворсин. Окраска гематоксилином и эозином.
Существенно отличалась плацента пациентки с преэклампсией: она имела нормальную массу 409 г (51 ‰), макроскопически число кистозных образований было небольшим. При гистологическом исследовании выявлено характерное для МДП увеличение размеров стволовых ворсин, отек их был умеренно выраженным, число сосудов небольшим. Морфология концевых ветвей была типична для преэклампсии, терминальные ветви представлены тонкими ветвящимися структурами с малочисленными узкими сосудами и большим числом синцитиальных узлов (см. рис. 2, е).
Строение и степень васкуляризации ворсин значительно различались как в разных отделах одной плаценты, так и между плацентами. В ряде случаев отмечены гигантские, извитые сосуды не только в хориальной пластинке, но и в стволовых ворсинах (см. рис. 2, ж). В других ворсинах имелись многочисленные узкие сосуды, нередко неправильной формы (см. рис. 2, з). Васкуляризация промежуточных и терминальных ворсин в большинстве случаев соответствовала норме, но в отдельных участках встречался хорангиоз. Экспрессия Ki-67 наблюдалась в немногочисленных клетках цитотрофобласта и стромы ворсин (см. рис. 2, и). Существенных особенностей экспрессии гладкомышечного актина и десмина в плацентах при МДП не выявлено.
Наблюдения подтвердили высокую частоту тромботических осложнений со стороны как матери, так и плода. В большинстве наблюдений в межворсинчатом пространстве был увеличен объем фибриноида, имелись тромбы разной давности (см. рис. 2, к). В четырех плацентах имелись признаки тромботической фетальной васкулопатии (см. рис. 2, л, м).

Экспрессия р57 в исследованных плацентах была вариабельной (рис. 3). Рис. 3. Варианты экспрессии р57 при МДП, иммуногистохимическая реакция. а — единичные позитивные клетки цитотрофобласта, ×100; б — позитивные клетки в цитотрофобласте и строме ворсин, ×200. В 3 наблюдениях реакция была положительной в клетках ворсинчатого цитотрофобласта при отсутствии экспрессии в строме ворсин. В 2 других наблюдениях экспрессия была очаговая: типичная для нормальной плацентарной ткани экспрессия р57 (окрашивание клеток цитотрофобласта и стромы ворсин) чередовалась с участками с полным отсутствием данного маркера. В одном наблюдении распределение р57-позитивных клеток соответствовало таковому в нормальной плаценте.
Заключение
Типичные морфологические признаки МДП: аномалия сосудов хориальной пластинки и стволовых ворсин, увеличение размеров и отек стволовых ворсин при нормальном формировании концевых ветвей ворсинчатого дерева, отсутствие пролиферации ворсинчатого трофобласта отмечены во всех наблюдениях. Плацентомегалия выявлена в 5 из 6 наблюдений. Плацента пациентки с преэклампсией и типичными ультразвуковыми и морфологическими признаками МДП имела также и существенные отличия от остальных изученных плацент. В данном наблюдении не выявлено плацентомегалии и отмечены характерные для преэклампсии изменения промежуточных и терминальных ворсин. Указанные особенности следует учитывать при анализе наблюдений МДП в сочетании с преэклампсией.
Сопоставление результатов ультразвукового исследования с морфологической картиной заболевания свидетельствует о наличии характерных для МДП ультразвуковых признаков, которые следует учитывать при дифференциальной диагностике с пузырным заносом.
Изменения показателей уровня хорионического гонадотропина и альфа-фетопротеина явились непостоянными признаками, таким образом не могут быть критериями для дифференциального диагноза МДП.
Высокая частота тромботических осложнений как в межворсинчатом пространстве, так и в фетальных сосудах, задержка внутриутробного развития плода, наличие внутриутробной гипоксии и нарушений периода адаптации новорожденных при МДП должны учитываться при определении тактики ведения пациенток и новорожденных.
Мы выявили значительную вариабельность экспрессии р57 при МДП, что ограничивает возможности определения данного маркера для диагностики МДП, а также косвенно свидетельствует о генетической гетерогенности указанной патологии. Следует настоятельно рекомендовать проведение генетического исследования при МДП в связи с данными о возможном наличии мозаицизма не только в плаценте, но и в тканях плода.
Синдром Беквита-Видеманна: укоротить язык, чтобы выжить

«Укоротить язык»: оказывается, так может звучать не только совет утомленных родителей маленьким болтунам, но и самое настоящее врачебное назначение. Макроглоссия, при которой язык вырастает так, что во рту не помещается, — один из симптомов синдрома Беквита-Видеманна, генетической аномалии, которая возникает без особых причин. Как ее диагностируют, что грозит малышам с большим языком, и можно ли предупредить заболевание, рассказывает MedAboutMe.
У 9 из 10 детей с синдромом причины заболевания неизвестны

Синдром гигантизма с пупочной грыжей, он же синдром Беквита-Видеманна — заболевание редкое. В среднем оно проявляется у одного из 13 000 младенцев. В 9 из 10 случаев никаких предпосылок к аномалии нет, дети с синдромом рождаются у здоровых родителей. Еще в 10% отмечают влияние наследственности: если у близких родственников есть такое же заболевание, шансы у ребёнка родиться с особенностями несколько выше. Если же синдром есть у матери или отца, то вероятность наследования — 50%.

Пока неясно, почему это происходит, но уже есть статистические данные: процедура ИКСИ, инъекционное оплодотворение сперматозоидами яйцеклетки, также повышает вероятность развития синдрома Беквита-Видеманна у ребёнка на фоне генетического здоровья родителей или доноров. Объяснения явлению еще нет, но у детей, родившихся после зачатия методом ИКСИ, синдром гигантизма с пупочной грыжей встречается несколько чаще, чем у остальных малышей.
У 8 из 10 детей с синдромом Беквита-Видеманна при генетическом анализе отмечают мутацию гена IGF-2 в 11-й паре хромосом. Этот ген называют инсулиноподобным фактором роста, а его изменение означает, что различные органы и ткани вдруг начинают расти неравномерно, опережая остальные по сроку развития. Среди вероятных мутаций — наличие только отцовского гена в обеих хромосомах или дублирование генов. А в 20% случаев мутацию вообще невозможно выявить, и диагноз ставят по симптомокомплексу болезни без подтверждения генетиком.
Гигантизм и пуповинная грыжа

Среди первых симптомов болезни, обнаруживаемых еще во время УЗИ, отмечают повышенные показатели веса и роста ребёнка, превышающие срок беременности. Иногда можно заметить, что показатели ассиметричны, например, отмечают увеличение внутренних органов: печени, селезенки, почек, нередко — языка.

Осиа Уорни, малышка с синдромом Беквита-Видеманна, показывала язычок окружающим еще во время ультразвукового исследования. Доктор и медсестра, наблюдавшие эту картину, нашли ситуацию «необыкновенно милой», а вот мама девочки встревожилась.
Осиа родилась некрупной, она — одна из пары близнецов, и у ее сестры Индиго никаких особенностей не отмечалось. После появления на свет девочке пришлось 7 дней провести в реанимации: помимо увеличенного язычка у нее выявили пониженный уровень сахара в крови, а это тоже один из первых симптомов заболевания.
Через 7 месяцев малышке провели первую операцию — пластику языка. Увеличенная мышца не помещалась во рту, мешала дышать, есть и учиться разговаривать. Сегодня Осиа полтора года, и она не отличается по здоровью от родной сестры, хотя каждые 6 месяцев ей приходится проходить обследование на онкофакторы: у людей с синдромом Беквита-Видеманна очень велики риски появления раковых опухолей.
Во время беременности макроглоссию, увеличенный язык, замечают довольно редко, да и сам этот симптом встречается не при каждом случае заболевания. Перинатальные признаки болезни — размеры плода, увеличенная пуповина, плацента, повышенное количество околоплодных вод.
После рождения, как правило, преждевременного, первым сигналом для наблюдения за ребёнком становятся показатели роста и веса. Крупный и недоношенный малыш — в группе риска. Гигантизм может быть, и чаще всего так и происходит, не полным. Могут быть увеличены внутренние органы, ушные раковины, язык. Однако все это в первые месяцы может проходить незамеченным или списываться на индивидуальные особенности ребёнка.
Тревожные сигналы — это низкий уровень глюкозы в крови, а также пуповинная грыжа, которая встречается настолько часто, что даже включена в название синдрома. Из-за порока развития брюшной стенки грыжа может быть достаточно большой: в пупочное кольцо и расходящийся диастаз выпадает часть кишечника, порой даже печень и другие органы пищеварения.
Среди дополнительных частых симптомов — гидронефроз, почечные кисты и почечная недостаточность, возможно наличие эмбриональных опухолей, микроцефалии, недоразвития лицевых мускулов и увеличенных глазных яблок.
Шансы на здоровье

Хотя в раннем детстве детишки с синдромом Беквита-Видеманна могут сильно отличаться от сверстников, выделяясь несоответствием размеров частей тела друг другу и в особенности высоким ростом, то к подростковому возрасту, как правило, эти особенности исчезают. Однако с рождения и как минимум до 8 лет дети переживают критический период развития — это специфика болезни.
Для коррекции размеров внутренних органов и вправления грыжи малышам нередко приходится проходить через ряд операций. Уменьшение размера языка тоже может происходить поэтапно.

Оливия Джиллис, маленькая подданная Великобритании, сегодня готовится в первый класс. Как и все британцы, она пойдет туда в 4 года. Девочка уже может нормально есть и говорить, хотя для этой возможности ей пришлось несколько раз побывать в операционной.
Оливия — один из ярких примеров малышей с макроглоссией, развившейся из-за синдрома Беквита-Видеманна. Сразу после рождения стало ясно, что операции не избежать. Язычок занимал все пространство во рту ребёнка, и кормить грудью малышку мама не смогла. Девочка получала молоко через трубку.
К полугоду ее язык не просто не помещался во рту: он полностью закрывал подбородок! В 6 месяцев ей провели первую операцию по коррекции формы, и ребёнок начал свободно дышать. Однако потребовалось еще две операции, чтобы вернуть языку малышки естественные пропорции.
Дополнительные опасности, которые приносит синдром Беквита-Видеманна, — это гипогликемия, пониженный уровень глюкозы, гипокальциемия, сниженный иммунитет и частые респираторные заболевания. А еще у детей очень высок риск развития онкологических опухолей, так, 7,5% случаев сопровождаются раком почек. Причем болезнь развивается быстро и агрессивно, поэтому все малыши и взрослые с синдромом должны регулярно проходить исследования.
Несмотря на сложности, взрослые пациенты отзываются о своем здоровье вполне позитивно. Они ведут полноценную жизнь, при которой основным напоминанием о заболевании является только необходимость несколько чаще посещать врачей для ранней диагностики возможных проблем.
Ребёнок проявляет беспокойство и жалуется на страшные сны? Не может сосредоточиться на занятии, волнуется и часто жалуется на больной живот? Наш тест поможет определить уровень тревожности у ребёнка и подскажет дальнейшую тактику поведения.
Читайте также:
- Рацемозная гемангиома сетчатки: признаки, гистология, лечение, прогноз
- Выбор метода родоразрешения при многоплодной беременности. Тактика родоразрешения при многоплодии.
- Механизмы возникновения субъективного шума в ухе. Сальпингоотит
- Тесты на сопротивление в голеностопном суставе и стопе
- Пациенты после пневмэктомии. Клиническое наблюдение пациентов после пневмэктомии