УЗИ при гистиоцитарном некротическом лимфадените (болезни Кикучи-Фуджимото)
Обновлено: 14.05.2024
Категории МКБ: Доброкачественное новообразование лимфатических узлов (D36.0), Другие неспецифические лимфадениты (I88.8), Неспецифический лимфаденит неуточненный (I88.9), Новообразование неопределенного или неизвестного характера лимфоидной, кроветворной и родственных им тканей неуточненное (D47.9), Отдельные болезни, протекающие с вовлечением лимфоретикулярной ткани и ретикулогистиоцитарной системы (D76), Саркоидоз лимфатических узлов (D86.1), Хронический лимфаденит, кроме брыжеечного (I88.1)
Общая информация
Краткое описание
Национальное гематологическое общество
НАЦИОНАЛЬНЫЕ КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ ПО ДИАГНОСТИКЕ ЛИМФАДЕНОПАТИЙ
2018
Лимфаденопатия (ЛАП) — увеличение лимфатических узлов любой природы в одной или нескольких анатомических зонах, относится к числу наиболее частых симптомов при многих заболеваниях и требует проведения тщательного диагностического поиска ее причины [1—7]. Этиология и патогенез ЛАП зависят от заболевания, симптомом которого она является.
Проблема дифференциального диагноза лимфаденопатий имеет большое значение для врачей многих специальностей: терапевтов, гематологов, онкологов, педиатров, хирургов, инфекционистов [8—11]. В большинстве случаев первичную диагностику проводит врач общей практики, направляющий больного к определенному специалисту. Приступая к диагностическому поиску, надо быть готовым к любому заболеванию [1—11].
Эффективный дифференциально-диагностический алгоритм предполагает знание по крайней мере основных причин увеличения лимфатических узлов. Основная проблема диагностики ЛАП состоит прежде всего в сходстве клинической картины опухолевых и неопухолевых ЛАП, которые занимают большое место в работе гематолога и онколога [9—11].
По данным исследования, проведенного в ФГБУ «НМИЦ гематологии», на неопухолевые ЛАП приходится 30% первичных обращений к гематологу по поводу увеличенных лимфатических узлов [5]. Результаты исследования 1000 больных с неопухолевыми ЛАП показали, что нозологический диагноз устанавливается всего в 50% случаев. Частота выполнения биопсий у больных с неопухолевыми лимфаденопатиями составила 46%. При этом только у трети подвергнутых биопсии больных с неопухолевыми ЛАП она имела решающее значение в постановке диагноза. В остальных случаях морфологическое исследование биопсированного лимфатического узла позволяло констатировать отсутствие опухоли, но не приводило к уточнению диагноза [5].
Спектры этиологической структуры при локальной и генерализованной ЛАП радикально отличаются. При локальной лимфаденопатии соотношение опухоль/не опухоль составляет 1:1 (48% и 52% соответственно), при генерализованной в 90% случаев выявляется опухоль и только в 10% подтверждается неопухолевый генез ЛАП [5]. У 10% больных с исходным диагнозом «неопухолевая лимфаденопатия» при повторной биопсии диагностируются опухоль или не классифицируемая редкая патология. Нередко разграничить лимфатическую опухоль и реактивный процесс не удается, особенно это касается диагностически трудных случаев, пограничных состояний, атипично протекающих лимфопролиферативных процессов. Диагноз в таких случаях устанавливается только со временем [5].
Анализ публикаций отечественных [1—5,8] и зарубежных [12—14] авторов, посвященных алгоритмам диагностики ЛАП, а также многолетний опыт работы «НМИЦ гематологии» [5] позволили разработать и внедрить протокол дифференциальной диагностики лимфаденопатий [15—17].
Лимфаденопатия - увеличение лимфатических узлов любой природы, относящееся к числу наиболее частых клинических симптомов, требующих проведения дифференциального диагноза.
D36.0 — доброкачественное новообразование лимфатических узлов (болезнь Кастлемана, локальные варианты);
D47.9 — новообразование неопределенного или неизвестного характера лимфоидной, кроветворной и родственных им тканей неуточненное;
D76 — отдельные болезни, протекающие с вовлечением лимфоретикулярной ткани и ретикулогистиоцитарной системы;
УЗИ при гистиоцитарном некротическом лимфадените (болезни Кикучи-Фуджимото)
Кафедра ЛОР-болезней, хирургии головы и шеи медицинского факультета Джеррахпаша Стамбульского университета
Редкое наблюдение шейной лимфаденопатии: болезнь Кикучи-Фуимото
Журнал: Вестник оториноларингологии. 2012;77(1): 65‑66
Yilmaz M., Mamanov M.A., Rashidov R., Ibrahimov M., Kyscu A., Karaman E. Редкое наблюдение шейной лимфаденопатии: болезнь Кикучи-Фуимото. Вестник оториноларингологии. 2012;77(1):65‑66.
Yilmaz M, Mamanov MA, Rashidov R, Ibrahimov M, Kyscu A, Karaman E. A rare case of cervical lymphadenopathy - Kikuchi-Fujimoto's disease. Vestnik Oto-Rino-Laringologii. 2012;77(1):65‑66. (In Russ.).
Болезнь Кикучи-Фуимото (БКФ) является формой гистиоцитарного некротизирующего лимфаденита, встречается преимущественно у женщин азиатского происхождения. Причина заболевания до конца не выявлена; предполагают иммунологическую и инфекционную природу заболевания. Клинически болезнь Кикучи-Фуимото проявляется обычно односторонней лимфаденопатией с лихорадкой и другими неспецифическими симптомами. Диагноз ставится на основе пункционной, чаще - эксизионной биопсии. Эффективной терапии для гистиоцитарного некротизирующего лимфаденита до сих пор не выявлено. Спонтанная регрессия может наступить в течение 1-6 мес. Представлено клиническое наблюдение больной 36 лет с верифицированной формой БКФ.
Болезнь Кикучи—Фуимото (БКФ) является формой гистиоцитарного некротизирующего лимфаденита и впервые была описана в Японии в 1972 г. Кикучи и Фуимото [1—3]. БКФ встречается преимущественно у женщин азиатского происхождения (соотношение женщины/мужчины 4:1) [1, 4, 5]. Пациенты могут быть в возрасте от 19 до 75 лет (чаще всего 25—29 лет).
Причина заболевания до конца не выявлена, но несколько клинических наблюдений позволяют предположить иммунологическую или инфекционную природу заболевания [6].
Клинически болезнь Кикучи—Фуимото проявляется обычно односторонней лимфаденопатией с лихорадкой и часто сочетается с другими неспецифическими симптомами, включающими гепатоспленомегалию, головную боль, похудание, недомогание, потливость ночью и желудочно-кишечные расстройства [7, 8].
Специфические радиологические характеристики не установлены. На УЗИ можно увидеть гипертрофированные лимфатические узлы с гипоэхогенным центром и гиперэхогенной окружностью. Лабораторные исследования также не имеют специфических показателей для постановки диагноза БКФ. В периферической крови больного выявляется лейкопения в 25,0—58,3% и в 25,0—31,1% случаев — атипические лимфоциты [9].

Диагноз ставится на основе пункционной, но чаще всего эксизионной биопсии [10]. Патологоанатомические данные имеют несколько особенностей: пораженные лимфатические узлы имеют регионарные очаги некроза (степень некроза варьирует) чаще всего в периаортальном пространстве. Очаги некроза содержат хорошо ограниченную зону эозинофильного фибриноидного материала с различной степенью выраженности ядерного разрыва (см. рисунок на цв. вклейке). Рисунок 1. Гистологическая картина образца ткани из шейного лимфатического узла больной У. Фокальный коагуляционный некроз, обломки ядра, гистиоциты, лимфоциты и большие клетки наподобие иммунобластов вокруг очага некроза. Окраска гематоксилином и эозином. Ув. 400.
Представляем редкое наблюдение болезни Кикучи—Фуимото.
Больная У., 36 лет, обратилась в ЛОР-кабинет поликлиники медицинского факультета Джеррахпаша Стамбульского университета 01.03.11 с жалобами на увеличение лимфатических узлов на шее, повышение температуры тела до 38,5 °С, похудание на 5 кг, незначительную общую слабость и недомогание в течение последних 4 нед.
При поступлении состояние средней тяжести. Сознание ясное. При наружном осмотре патологии не выявлено, кожные покровы обычной окраски, тургор не изменен. Патологических изменений в костно-мышечной и суставной системах не выявлено. В легких жесткое дыхание, сухие хрипы на выдохе, ЧД 22 в минуту. Тоны сердца приглушены, ритмичные, ЧСС 80 в минуту, АД 130/80 мм рт.ст. Живот мягкий, безболезненный. Печень по краю реберной дуги. Периферических отеков нет.
При осмотре ЛОР-органов патологии не выявлено, носовое дыхание не затруднено. При пальпации шеи в правой и левой боковых областях были обнаружены несколько небольших незначительно болезненных подвижных неспаянных лимфатических узлов.
УЗИ шеи показало увеличенные лимфатические узлы справа 12×5 мм, в левой шейной области 12×7 мм. Общий анализ крови от 02.03.11: л. 4,93·10 9 /л, э. 1,4, лимф. 53, мон. 11; СОЭ 40 мм/ч. Больной был назначен курс антибиотикотерапии. После курса лечения в состоянии больной и в размерах лимфатических узлов существенных изменений не произошло. Было решено выполнить аспирационную биопсию лимфатических узлов. При исследовании материала не было установлено точного патологического диагноза, но не исключалась лимфома Ходжкина. Для постановки окончательного диагноза была проведена эксизионная биопсия. В результате был верифицирован редко встречающийся гистиоцитарный некротический лимфаденит — лимфаденит Кикучи— Фуимото.
Больной было назначено лечение нестероидными противовоспалительными препаратами и преднизолоном (доза преднизолона 1 мг/кг, через каждые 2 дня уменьшали дозу на 10 мг) в течение 14 дней. При контрольном осмотре через 30 дней после лечения было отмечено значительное уменьшение шейных лимфатических узлов и понижение температуры тела до нормальных цифр.
Особенностью данного наблюдения является то, что такую редкую патологию, как гистиоцитарный некротизирующий лимфаденит (болезнь Кикучи—Фуимото), можно легко спутать с такими патологиями, как лимфома, туберкулез, гранулематоз Вегенера и с другими инфекционными заболеваниями.
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России
ФГБОУ ВО «Северо-Западный государственный медицинский университет им. И.И. Мечникова» Минздрава России;
СПбГБУЗ «Городская больница №40»
Анализ фенотипической гетерогенности CD123-позитивных клеток при болезни Кикучи—Фуджимото с помощью метода последовательного иммунопероксидазного окрашивания и стирания
Журнал: Архив патологии. 2021;83(4): 36‑44
Болезнь Кикучи—Фуджимото (БКФ) — редкое заболевание, которое клинически проявляется преимущественно лихорадкой и лимфаденопатией. Первоначально предполагалось, что БКФ встречается в основном у женщин в Восточной Азии, впоследствии это заболевание было описано во всех этнических группах по всему миру. Важным дифференциально-диагностическим признаком БКФ считается обнаружение в ткани пораженного лимфатического узла плазмоцитоидных дендритных клеток (ПДК), которые экспрессируют CD123. Стандартный иммуногистохимический метод окрашивания обладает достаточной чувствительностью и специфичностью для выявления CD123, но не позволяет судить о возможной фенотипической гетерогенности клеток с экспрессией CD123.
ЦЕЛЬ ИССЛЕДОВАНИЯ
Выявить фенотипическую гетерогенность клеток, экспрессирующих CD123, в пораженных лимфатических узлах у пациентов с БКФ методом последовательного иммунопероксидазного окрашивания и стирания (SIMPLE).
МАТЕРИАЛ И МЕТОДЫ
Исследованы эксцизионные биопсии лимфатического узла у 3 пациентов с БКФ. После выполнения иммуногистохимической реакции с одним антителом гистологический препарат переводили в цифровой формат с помощью сканера Pannoramic 250 Flash III (3DHISTECH, Венгрия), далее со среза снимали покровное стекло, гидратировали и помещали в специализированный буфер. Затем на промытый гистологический препарат наносили следующее первичное антитело и выполняли дальнейшую иммуногистохимическую реакцию и сканирование. В результате каждый гистологический препарат последовательно был окрашен в реакциях с 4 антителами. На микрофотографиях препаратов, окрашенных в реакции с CD123-антителом, отмечали позитивные клетки для их идентификации в программе Pannoramic Viewer («3DHISTECH», Венгрия) на остальных микрофотографиях, демонстрирующих экспрессию других 3 маркеров. Выбранные поля зрения экспортировали в JPG-формат.
РЕЗУЛЬТАТЫ
На основе оценки коэкспрессии антигенов CD123, MNDA, CD68, TCL1A были обнаружены 4 субпопуляции CD123+-клеток: №1: CD68+/ MNDA+/ TCL1A+; №2: CD68+/ MNDA+/ TCL1A-; №3: CD68+/ MNDA-/ TCL1A+; №4: CD68-/ MNDA-/ TCL1A+.
ЗАКЛЮЧЕНИЕ
Метод последовательного иммунопероксидазного окрашивания и стирания показал фенотипическую гетерогенность CD123-позитивных клеток (часть из них, вероятно, ПДК) и позволил выделить 4 иммунофенотипически различные субпопуляции в пораженных лимфатических узлах у пациентов с БКФ. Необходимы дальнейшие исследования для определения роли субпопуляций в патогенезе БКФ и других заболеваний.
Дата принятия в печать:
Болезнь Кикучи—Фуджимото (гистиоцитарный некротизирующий лимфаденит) — редкий патологический процесс, который характеризуется подострой некротизирующей регионарной лимфаденопатией, лихорадкой, ночной потливостью и часто ошибочно диагностируется как лимфопролиферативное заболевание, туберкулез или лимфаденит при системной красной волчанке [1].
Впервые гистиоцитарный некротизирующий лимфаденит был описан M. Kikuchi и Y. Fujimoto в 1972 г. [2, 3]. Заболевают лица молодого возраста (средний возраст пациентов 27,4 года, соотношение мужчин и женщин 1:1,6) [4—6]. Болезнь Кикучи—Фуджимото (БКФ) обычно манифестирует шейной лимфаденопатией (83% больных), реже поражаются подмышечные, паховые лимфатические узлы и лимфатические узлы средостения [7]. Известны единичные случаи экстранодальной локализации с вовлечением в патологический процесс печени, селезенки, кожи. Пораженные лимфатические узлы безболезненные при пальпации, а регионарная лимфаденопатия может сопровождаться умеренной лихорадкой, ознобом, миалгией, тонзиллитом и кожной сыпью, что в некоторых случаях требует проведения дифференциальной диагностики с корью и крапивницей. В 25% случаев клиническое течение БКФ у больных сопровождается анемией, нейтропенией, лимфоцитозом. Тем не менее БКФ — доброкачественное заболевание с хорошим прогнозом, саморазрешение болезни наступает в течение нескольких недель, рецидивы редки [4, 8].
Этиопатогенез БКФ изучен недостаточно. Предполагается, что вирусные инфекции (вирус Эпштейна—Барр, цитомегаловирусная инфекция, парвовирус B19, вирус герпеса человека 6-го типа и др.) могут выступать в роли триггерных факторов, способствующих манифестации заболевания [9—11]. K. Ohshima и соавт. [12], показали, что в острой стадии БКФ повышена сывороточная концентрация гамма-интерферона, интерлейкина-6 (ИЛ-6) и других провоспалительных цитокинов. При гистологическом исследовании биопсийного и операционного материала в пораженных лимфатических узлах находят очаги паракортикальных коагуляционных некрозов неправильной формы с кариорексисом гистиоцитов и макрофагальной инфильтрацией. В очаге поражения отсутствуют эозинофильные и нейтрофильные гранулоциты. Капсула лимфатического узла утолщена, рисунок строения частично стерт. Очаги некрозов окружены скоплениями гистиоцитов, иммунобластов, плазмоцитоидных дендритных клеток (ПДК) и малых лимфоцитов. Такая гистологическая картина может быть ошибочно интерпретирована как лимфома или миелоидная саркома. T. Kuo [5] выделил три стадии БКФ: пролиферативную, некротизирующую и ксантоматозную. Пролиферативная стадия характеризуется гиперплазией паракортикальной зоны с большим количеством гистиоцитов и ПДК. При некротизирующей стадии в пораженном лимфатическом узле появляются очаги некроза разной степени протяженности. Некротизирующая стадия может сменять пролиферативную. Если при гистологическом исследовании пораженного лимфатического узла видны скопления пенистых гистиоцитов независимо от наличия и отсутствия участков некроза, то такое состояние предлагают трактовать как ксантоматозная стадию.
Морфологическая диагностика БКФ предполагает обнаружение ПДК, они среднего размера, с округлым ядром и мелкодисперсной структурой хроматина, амфофильной цитоплазмой. ПДК обычно находят в лимфатическом узле в виде скоплений вне зон некроза (рис. 1). В ряде работ описана «атипичная» морфология ПДК при БКФ, что в сочетании с измененной гистоархитектоникой лимфатического узла требует дифференциальной диагностики с лимфомой высокой гистологической степени злокачественности [13, 14].

Рис. 1. Фрагмент лимфатического узла при болезни Кикучи—Фуджимото.
а — фокус некроза с кариорексисом (справа), ×230; б — клеточный состав гистиоцитарного некротизирующего лимфаденита. Окраска гематоксилином и эозином, ×330.
Важный иммуногистохимический маркер для выявления ПДК — цитоплазматическая экспрессия α-цепи рецептора ИЛ-3 (CD123) [15]. Также эти клетки экспрессируют CD68, CD303 и не экспрессируют миелопероксидазу (рис. 2).

Рис. 2. Иммуногистохимические маркеры, характерные для плазмоцитоидных дендритных клеток.
а — окраска гематоксилином и эозином; б — CD123; в — MNDA; г — TCL1A, ИГХ-окрашивание по методу SIMPLE, ×450.
Попытки изучить происхождение и функцию ПДК были предприняты исследователями в разное время [16—18]. Известно, что с помощью toll-подобных рецепторов ПДК способны распознавать вирусные ДНК и РНК, продуцировать интерфероны I и III типов [9]. ПДК выполняют функцию в работе противовирусного иммунитета, реализации патологических аутоиммунных реакций [10]. Несмотря на диагностическую значимость ПДК, их иммунофенотип при БКФ изучен недостаточно, что и предопределило цель работы.
Цель исследования — выявить фенотипическую гетерогенность субпопуляций CD123-позитивных клеток в лимфатических узлах у больных гистиоцитарным некротизирующим лимфаденитом (болезнь Кикучи—Фуджимото) методом последовательного иммунопероксидазного окрашивания и стирания (A Sequential Immunoperoxidase Labeling and Erasing Method — SIMPLE).
Материал и методы
Исследованы лимфатические узлы, полученные от 3 больных с клинически и гистологически подтвержденным диагнозом БКФ. Из парафиновых блоков на ротационном микротоме изготавливали срезы толщиной 3—4 мкм. При иммуногистохимическом окрашивании использовали первичные антитела фирм «DAKO» (Дания) и «Thermo Scientific» (США). Для визуализации антител применяли систему EnVision («DAKO», Дания) или Quanto («Thermo Scientific», США). Проявление пероксидазной метки осуществляли хромогеном Vector NovaRed («Vector laboratories», США). Докрашивание гематоксилином, дегидратация и заключение под бальзам гистологических препаратов выполняли по стандартной методике. Полученные гистологические препараты переводили в цифровой формат с помощью лабораторного сканирующего микроскопа (сканера) Pannoramic 250 Flash III («3DHISTECH», Венгрия). После получения оцифрованных изображений с гистологических препаратов снимали покровное стекло, гидратировали их и помещали в буфер, содержащий 2% SDS, 0,8% β-меркаптоэтанол и 62,5 ммоль/л трис-HCl; pH 7,5. На промытые в TBS гистологические препараты наносили следующие по списку первичные антитела (см. таблицу), дальнейшее окрашивание среза выполняли по методике, приведенной выше.
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Научная группа при кафедре болезней уха, горла и носа Первого МГМУ им. И.М. Сеченова Минздравсоцразвития России
Кафедра оториноларингологии педиатрического факультета Российского национального исследовательского медицинского университета им. Н.И. Пирогова, Москва, Россия, 117997
Морозовская ДГКБ Департамента здравоохранения Москвы, Москва, Россия, 119049
Болезнь Розаи—Дорфмана: обзор литературы и клиническое наблюдение экстранодальной формы заболевания с поражением слизистой оболочки носа и околоносовых пазух
Журнал: Вестник оториноларингологии. 2018;83(6): 72‑75
Синусный гистиоцитоз с массивной лимфаденопатией (болезнь Розаи—Дорфмана) представляет собой редкое неопухолевое заболевание неизвестной этиологии с доброкачественным течением. В статье представлены обобщенные данные литературы и клиническое наблюдение экстранодальной формы заболевания с поражением слизистой оболочки носа и околоносовых пазух.
Описанный впервые в 1969 г. J. Rosai и R. Dorfman [1] синусный гистиоцитоз с массивной лимфаденопатией (СГМЛ) — болезнь Розаи—Дорфмана представляет собой редкое неизвестной этиологии доброкачественное неопухолевое заболевание, характеризующееся накоплением пролиферирующих гистиоцитов в синусах лимфатических узлов, в результате чего происходит их массивное увеличение. СГМЛ относится к группе так называемых заболеваний с атипичными клеточными нарушениями лимфатических узлов. Атипия в данном случае проявляется тем, что ненормальное увеличение количества гистиоцитов происходит в результате выраженного фагоцитоза ими клеток лимфоидного ряда. Редкость заболевания, противоречивость различных описаний, неопределенность специфических гистиоцитарных маркеров и четких методов определения клональности, а также существование ряда близких клинических и гистологических состояний реактивного инфекционного и опухолевого происхождения делают это заболевание сложным для диагностики. По данным Гематологического научного центра РАМН, более чем у 50% больных с синусным гистиоцитозом в дальнейшем выявляются онкогематологические заболевания [2].
Этиология СГМЛ остается неизвестной, и хотя его клинические проявления и гистологическая картина сходны с наблюдаемыми при инфекционных процессах, микроорганизмы при СГМЛ до настоящего времени не идентифицированы [3]. Предполагают, что в патогенезе СГМЛ имеют значение повсеместно распространенные вирус герпеса 6-го типа и вирус Эпштейна—Барр, которых обнаруживают более чем у 50% больных СГМЛ [3, 4]. Данные молекулярных исследований указывают на то, что СГМЛ — поликлональное заболевание [5].
После создания в 1990 г. регистра пациентов, страдающих СГМЛ, выяснилось, что клинические признаки достаточно разнообразны: если в первых описаниях упоминалось заболевание с выраженной двусторонней лимфаденопатией с поражением преимущественно шейных узлов у маленьких детей негроидной расы [1, 6], то анализ регистра показал, что возраст больных варьирует от новорожденности до 74 лет, мужчины заболевают в 1,5 раза чаще женщин, наряду с «излюбленной» локализацией в шейных лимфоузлах могут быть поражены и другие группы лимфатических узлов, а у 40% больных выявляют экстранодальные очаги поражения [3]. В большинстве случаев локализованная лимфаденопатия является первым и единственным проявлением заболевания, хотя описаны случаи СГМЛ в виде псевдоопухолей на коже, орбите, в среднем ухе, верхних дыхательных путях, желудочно-кишечном тракте, мозговых оболочках и др. [2, 7—14].
Чаще заболевание имеет доброкачественное течение, склонное к спонтанной регрессии, сопровождающееся частыми рецидивами, однако лимфаденит может персистировать годами. Клиническая картина СГМЛ характеризуется массивной лимфаденопатией с преимущественным поражением шейных лимфатических узлов, отсутствием симптомов интоксикации. Помимо увеличения лимфатических узлов и экстранодальных очагов, при обострении наблюдается лихорадка, лейкоцитоз, повышение СОЭ. У ряда больных отмечаются нарушения уровня сывороточных белков в виде умеренной поликлональной гипергаммаглобулинемии. У части пациентов встречаются те или иные признаки поражения иммунной системы: полиартралгия, бронхолегочные поражения или гемолитическая анемия, предшествующие развитию СГМЛ или развивающиеся в дебюте [2, 15].
Диагноз устанавливается исключительно на основании морфологического исследования биопсийного материала. Микроскопически структура лимфатического узла обычно нарушена за счет резко выраженного расширения синусов, стертости фолликулов и герминальных центров. Характерными признаками являются склерозирование капсулы и перикапсулярное разрастание соединительнотканных волокон и жировой ткани. Растянутые синусы и медуллярные тяжи содержат смешанную популяцию клеток, представленную полиморфно-клеточными лейкоцитами и лимфоцитами, однако преобладающей популяцией являются гистиоциты, характеризующиеся выраженным полиморфизмом. Это крупные клетки с большой светлой, иногда вакуолизированной цитоплазмой с одним или несколькими вогнутыми ядрами. Митозы наблюдаются редко. Внутрицитоплазменные вакуоли содержат фагоцитированные лимфоциты, эритроциты или нейтрофилы, они не подвергаются воздействию цитолитических ферментов. Некоторые клетки, особенно лимфоциты, способны жить в вакуолях (феномен эмпериополеза), другие постепенно деградируют, формируя ядерные фрагменты. Некоторые вакуоли содержат только остатки ядер деградировавших клеток или липиды, которые хорошо окрашиваются суданом. В мозговом слое отмечаются многочисленные плазматические клетки, часть из которых двуядерные. Эозинофилы встречаются редко. Некроза также не отмечается. Микроскопическая картина экстранодальных очагов СГМЛ сходна с наблюдаемой в лимфатических узлах, хотя экстранодальные очаги характеризуются более выраженным фиброзом, менее выраженным скоплением гистиоцитов и лимфоцитофагоцитозом [2].
Цитологические препараты лимфатического узла гиперклеточны, с обилием гистиоцитов, фагоцитированных лимфоцитов на фоне реактивации лимфоидной ткани. Гистиоциты обычно с рыхлым ядром и обильной бледной, часто вакуолизированной цитоплазмой и феноменом эмпериополеза. Атипичные гистиоциты содержат гиперхромные ядрышки, некоторые гистиоциты достигают гигантских размеров. На ранних стадиях СГМЛ в мазках можно увидеть множество лимфоцитов и иногда иммунобласты [16]. На более поздних стадиях доминируют многочисленные плазматические клетки и тельца Русселя [17]. В большинстве случаев имеется лизосомная активность гистиоцитов, хотя число лизосом может варьировать в широких пределах, вплоть до их отсутствия. При иммуногистохимическом исследовании установлена принадлежность клеток при СГМЛ к макрофагально-гистиоцитарной группе. Но неясно, к какому конкретно представителю этой группы они относятся, так как во всех случаях СГМЛ происходит экспрессия белка S100, который является специфическим маркером интердигитирующих клеток в лимфатических узлах и клеток Лангерганса в коже [18].
Дифференциальный диагноз необходимо проводить со следующими заболеваниями: синусный гистиоцитоз как неспецифическая реакция лимфатических узлов на инфекцию или опухоль, злокачественный гистиоцитоз, гранулематозное поражение; эозинофильная гранулема; болезнь Хенда—Шюллера—Крисчена (для нее характерно поражение скелета), болезнь Леттерера—Сиве (встречается у младенцев, характерны поражения кожи), болезнь Ходжкина [2, 3, 14].
В настоящее время методы лечения СГМЛ не разработаны [14, 19]. В большинстве случаев прогноз благоприятный [8].
В связи с редкостью болезни Розаи—Дорфмана считаем целесообразным привести казуистическое наблюдение экстранодальной ее формы с поражением слизистой оболочки носа и околоносовых пазух.
Родители мальчика К., 4 лет (2013 г. р.), впервые обратились в Морозовскую ДГКБ в сентябре 2016 г. с жалобами на наличие образования на конъюнктиве правого глаза. Офтальмологом обнаружен сосудистый невус конъюнктивы склеры правого глаза, рекомендовано местное лечение, динамическое наблюдение, при отсутствии положительной динамики — плановое хирургическое лечение. Через 3 мес после первичного обращения, в декабре 2016 г., родители обнаружили образование в области левого яичка. В связи с начавшимся через 4 мес после обнаружения ростом образования яичка (апрель 2017 г.) по месту жительства была выполнена МРТ: согласно заключению, картину необходимо дифференцировать между сосудистой опухолью и гамартомой левой половины мошонки. Лабораторные исследования крови на онкомаркеры: определение уровня АФП — 0,135 мМЕ/мл и β-ХГЧ — 0,92 МЕ/мл, убедительных данных за онкологический процесс не получено. Выполнено УЗИ мошонки (апрель 2017 г.): левое яичко в мошонке размером 1,0×0,7×0,7 см, выше него определяется образование узлового характера неоднородной структуры размером 0,8×0,6×1,5 см, рядом с ним расположены еще несколько узлов.
Ребенок консультирован онкологом Морозовской ДГКБ — рекомендовано удаление образования со срочным гистологическим исследованием и решением вопроса об объеме операции по результатам интраоперационного гистологического ответа. В июле 2017 г. выполнена поперечная скрототомия — при ревизии мошонки обнаружено объемное образование, плотное, желтоватого цвета, с неровными контурами, интимно прилежащее к нижнему полюсу левого яичка, размером 2×3 см. Дистальнее, по ходу элементов семенного канатика, обнаружено подобное образование размером 0,8×0,5 см. Удаленные объемные образования направлены на экспресс-биопсию. Интраоперационно получен ответ — рабдомиосаркома, в связи с чем онкологом было рекомендовано выполнение орхифуникулэктомии паховым доступом. Однако при дальнейшем гистологическом и иммуногистохимическом исследовании удаленного образования и регионарных лимфатических узлов у ребенка установлен диагноз синусного гистиоцитоза — болезни Розаи—Дорфмана. В дальнейшем рекомендованы консультация и наблюдение в центре детской гематологии и онкологии Морозовской ДГКБ.
В сентябре 2017 г. родители обратились к оториноларингологам Морозовской ДГКБ в связи жалобами на затруднение носового дыхания в течение нескольких месяцев. При осмотре — носовое дыхание через левую половину носа отсутствует, справа — свободное. При диагностической фиброэндоскопии полости носа и носоглотки обнаружено, что поверхность носовых раковин покрыта видоизмененной бугристой слизистой оболочкой бледно-розового, местами желтоватого цвета, при надавливании на которую появляется вязкое слизистое отделяемое. Через левую половину носа доступ к носоглотке невозможен в связи с наличием плотного, розово-серого цвета образования, исходящего из левого среднего носового хода, из левой верхнечелюстной пазухи и блокирующего левую половину носа в средних отделах; не кровоточащего при пальпации. Слизистая оболочка (СО) носовых раковин справа аналогично изменена, общий носовой ход достаточно широкий, хоана обычной формы, в куполе носоглотки лимфоидная ткань на уровне ½ сошника, трубные валики не увеличены.
Пациенту выполнена компьютерная томография полости носа и околоносовых пазух с контрастным усилением (ультравист) в условиях аппаратно-масочной анестезии (сентябрь 2017 г.) — отмечается тотальное заполнение патологическим содержимым левой верхнечелюстной пазухи, субтотальное заполнение клеток левого решетчатого лабиринта; медиальная стенка левой верхнечелюстной пазухи частично разрушена. Отмечается наличие периостальных наслоений по внутренней поверхности стенок пазухи. СО правой верхнечелюстной пазухи неравномерно утолщена, СО носовых раковин утолщена (рис. 1, 2). Рис. 2. Пациент К. Компьютерная томограмма полости носа и околоносовых пазух в мягкотканном режиме. а — аксиальная; б — коронарная проекции. Рис. 1. Пациент К. Компьютерная томограмма полости носа и околоносовых пазух в костном режиме. а — аксиальная; б — коронарная; в — сагиттальная проекции.
Ребенок консультирован онкологом (сентябрь 2017 г.), которым высказано предположение об экстранодальном проявлении болезни Розаи—Дорфмана. Рекомендовано удаление образования.
После предоперационной подготовки под интубационным наркозом выполнена левосторонняя эндоназальная эндоскопическая этмоидогайморотомия, удалены патологическое образование из верхнечелюстной пазухи, а также патологически измененная СО нижних носовых раковин и клеток решетчатого лабиринта с обеих сторон.
По данным патоморфологического исследования операционного материала (гистологическое и иммуногистохимическое исследование; окраска IHC: CD1a, CD3, CD38, CD68, CD79a, Macrophage, S100) определяются механически деформированные фрагменты СО околоносовых пазух с мелким участком костной ткани, частично покрытые многорядным призматическим реснитчатым эпителием, в собственной пластинке СО отек, очаговый склероз, диффузная инфильтрация большого количества крупных гистиоцитов с обильной вакуолизированной цитоплазмой, округлыми ядрами с крупным хроматином и различимым одним ядрышком и явлениями эмпериополеза (фагоцитированные лимфоциты, эритроциты и плазматические клетки). Тотальная экспрессия в воспалительном инфильтрате S100, CD68, Macrofage, CD79a, CD3, CD38. Реакция отрицательна с антителом к CD1a. Заключение: морфологическая картина соответствует болезни Розаи—Дорфмана, экстранодальный вариант.
Также проведено исследование на базе ФГБУ «Гематологический научный центр» Минздрава России.
Микроскопическое исследование: мелкие фрагменты СО, покрытой многоядерным мерцательным эпителием со слизистыми железами: фрагменты рыхлой соединительной ткани с пролифератом, представленным крупными скоплениями зрелых плазматических клеток, инфильтрацией из мелких лимфоидных клеток, макрофагов, наличием крупных гистиоидных клеток с пузырьковидным ядром, светлой/слабоэозинофильной широкой цитоплазмой, отдельные из них — с признаками эмпериополеза.
При иммуногистохимическом исследовании препаратов: крупные гистиоидные клетки с пузырьковидными ядрами, расположенные разрозненно и в виде рыхлых скоплений среди лимфоплазмоцитарного инфильтрата с примесью макрофагов, экспрессируют S-100, ядерно-цитоплазматическая реакция СD68, многочисленные зрелые плазматические клетки CD38 — позитивны. Реакция с антителом к CD7 позитивна в мелких В-клетках, в зрелых плазматических клетках многочисленная Т-клеточная популяция CD3+. Заключение: морфологическая картина и иммунофенотип характеризуют нелангергансоклеточный гистиоцитоз — экстранодальный субстрат болезни Розаи—Дорфмана (клональную гистиоцитарную пролиферацию).
В послеоперационном периоде производился туалет полости носа растворами антисептиков, в общих носовых ходах были установлены тампоны, которые были удалены на 5-е сутки, и на 7-е сутки ребенок выписан домой в удовлетворительном состоянии.
За истекший период — 6 мес рецидива данного процесса не отмечено.
Заключение
Болезнь Розаи—Дорфмана представляет собой одно из редких заболеваний неизвестной пока этиологии, характеризующееся выраженным синусным гистиоцитозом. Диагноз в данном случае был установлен на основании классических гистологических данных, в том числе явлений эмпериополеза, типичной иммуногистохимической характеристики гистиоцитов в синусах.
Особенностью данного случая является экстранодальное поражение СО носа и околоносовых пазух с частичным разрушением костных структур, сопровождавшееся яркими клиническими проявлениями. В связи с отсутствием разработанных методов химиотерапевтического воздействия особую важность в диагностике и терапии имеет правильное и эффективное хирургическое лечение.
Гистиоцитарный некротический лимфаденит на УЗИ
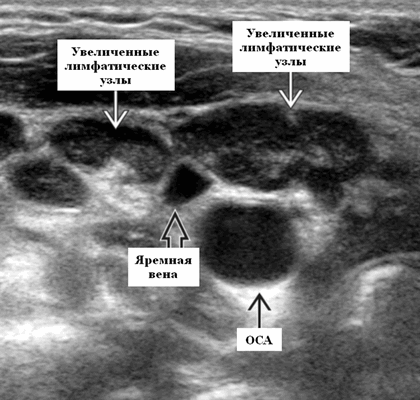
Терминология. Сокращения. Гистиоцитарный некротический лимфаденит (ГНЛ). Синонимы. Болезнь Кикучи. Болезнь Кикучи-Фуджимото
Определение. Некротический лимфаденит с преобладающей гистиоцитарной инфильтрацией
Основные характеристики при ультразвуковом сканировании
- Лучший диагностический признак на УЗИ - односторонние, овальные / эллиптические, слегка увеличенные, гипоэхогенные, дискретные лимфатические узлы (уровни II-V) у молодых азиатских пациенток
- Расположение. Задний треугольник шеи или внутренняя яремная цепь. Большинство на уровнях II и V. Односторонние изменения встречаются чаще, чем двусторонние
- Размер. Обычно 0,5-4,0 см (в среднем: 1,6 см), меньше лимфомных узлов
- Рекомендации по визуализации. Лучший инструмент для диагностики - сонография лучше всего показывает некроз, также сразу можно провести тонкоигольную биопсию под контролем УЗИ
Ультрасонографические данные
Результаты КТ. Переменные изображения отображаются в зависимости от степени некроза. Однородно усиливающиеся, сплошные узлы. Измененные, центрально гиподенсивные узлы. Перинодальные активные изменения, который предлагает воспалительный процесс
МРТ данные
• T2WI. Центральные, неусиливающиеся, некротически выглядящие области не гиперинтенсивные
• T1WI C + FS. Солидно усиливающие или усиливающие периферию лимфоузлов
Результаты ядерной медицины. ПЭТ / КТ. Увеличенное поглощение FDG в увеличенных узлах. Имитирует неходжкинскую лимфому (НХЛ)

Дифференциальная диагностика при ультразвуковом сканировании
- Реактивные лимфатические узлы. Похожи на узлы неходжкинской лимфоме, часто имеют двусторонний и симметричный характер. При лимфоме обычно имеется длительное клиническое течение
- Туберкулезные лимфатические узлы. Некроз является ранним и распространенным явлением; смещенная внутригрудная сосудистая сеть. Матирование и воротниковый абсцесс ± интранодальная кальцификация в узлах после лечения
- Узлы неходжкинской лимфомы. Группа пожилых пациентов; большие, более круглые лимфоузлы ан УЗИ. Большинство с ретикулярным или псевдосолидным рисунком. Смешанная сосудистая система, интранодальный кровоток более выражен, чем периферический
- Системные узловые метастазы. Круглые сплошные лимфоузлы с отсутствием ворот, ± интранодальный некроз ± кальцификация. Периферический сосудистый рисунок и известный первичный опухолевый очаг
- Болезнь кошачьих царапин. Региональная аденопатия после царапины. Рукавовлекается чаще, чем шея и обычно встречается у детей и подростков
Клинические проблемы и проявления болезни Кикучи-Фуджимото
- Наиболее распространенные признаки / симптомы. Острая подострая болезненная шейная лимфаденопатия. Около 90% включают задний треугольник и примерно 90% односторонние изменения на УЗИ. Размер лимфатического узла в большинстве случаев составляет 0,5-4,0 см. 30-50% пациентов имеют низкую температуру. Верхний дыхательный продром. Лабораторный тест обычно нормальный. Больной может иметь лейкопению, повышенное СОЭ, сывороточную ЛДГ и АСТ, атипичные лимфоциты периферической крови
- Другие признаки и симптомы. Генерализованная лимфаденопатия (1-22%). Потеря веса, тошнота, рвота, ночные поты. Пирексия неизвестного происхождения. Наружные проявления кожи (30-50%). При осмотре поражение кожи или костного мозга, гепатоспленомегалия, дисфункция печени. Кожное проявление чаще встречается у пациентов мужского пола. Широкий спектр дерматологических паттернов от сыпи до макулопапулезных высыпаний. В основном влияет на лицо и верхнюю часть тела
Демография
- • Возраст. Молодые люди, средний возраст 25 лет
- • Пол. Болезнь преобладает у взрослых женщин. Женщины: Мужчины = 1-2: 1. У детей Мальчики> Девочки
- • Эпидемиология. Высокая распространенность среди азиатских наций, особенно японских пациентов
- Естественная история и прогноз
- • Доброкачественная, самоограниченная болезнь. Длится 1-4 месяца. Спонтанное разрешение аденопатии
- • Низкая частота рецидивов 3-4%
- • Редко сложный курс с плохим результатом. Чаще встречается при экстранодальном поражении. Легочное кровотечение, сердечная недостаточность, фатальный гемофагоцитарный синдром
Лечение. Симптоматическая: анальгетики, жаропонижающие, отдых
Итог
Читайте также: