Синдром Линдау (Lindau) - синонимы, авторы, клиника
Обновлено: 15.05.2024
Гиппеля-Линдау болезнь (E. Hippel, немецкий офтальмолог, 1867—1939; А. V. Lindau, шведский патолог, 1892—1958; син.: множественный ангиоретикулематоз, ретиноцеребровисцеральный ангиоматоз, семейный ангиоматоз) — заболевание, характеризующееся развитием ангиоматозных, ангиоретикулематозных и кистозных образований в сетчатке глаз, центральной нервной системе и внутренних органах. Относится к группе факоматозов (см.).
Офтальмологическая симптоматика была впервые описана Панасом (Ph. Panas, 1879) и Реми (A. Remy, 1892), а затем детально Гиппелем в 1895, 1903, 1904 гг. После классических исследований Линдау в 1926—1927 гг., доказавшего системность этого заболевания и описавшего церебральные нарушения, Гиппеля-Линдау болезнь вошла в медицину как самостоятельная нозологическая форма. Болезнь встречается относительно редко.
Содержание
Этиология и патогенез
Заболевание является наследственным. С. Н. Давиденков (1958), И. С. Бабчин (1962), Шокейр (Shokeir, 1970) и другие считают, что ангиомы сетчатки, мозга и гипернефромы имеют единый онкологический генез. Прогредиентность, сочетание ангиоматоза с опухолевой клеточной структурой, семейный характер — доказательства единой природы болезни. В связи с частотой семейных форм следует говорить не о наследственном предрасположении, а о генетически детерминированной болезни с довольно стойкой пенетрантностью гена и с незначительным влиянием других генотипических факторов и окружающей среды. Подтверждением служит наблюдаемый в нескольких поколениях одинаковый возраст к началу заболевания, однотипность форм, локализации ангиоматозных разрастаний и сочетания определенных аномалий развития скелета, эндокринной системы, внутренних органов. Заболевание наследуется преимущественно по аутосомно-доминантному типу.
Патологическая анатомия
В основе патологического процесса Гиппеля-Линдау болезни лежит развитие капиллярных ангиом сетчатки глаза, в дальнейшем — формирование кист, артериовенозных аневризм со вторичным разрастанием глии. В сетчатке уже в начальном периоде болезни обнаруживают помутнение, расширение и извитость сосудов, преимущественно капилляров. Ангиомы развиваются чаще всего в экваториальной части, и в первую очередь в зоне капилляров между артериальными и венозными стволами. Позже артерии и вены становятся утолщенными, извитыми, расширенными, разрастающиеся капилляры образуют клубочки ярко-красного цвета с желтым оттенком, в которых затем появляется экссудат, геморрагии. Прогрессирование процесса ведет к отслойке сетчатки (см.), атрофии зрительного нерва (см.). Гистологически в протоплазме опухолевых клеток между тяжами капилляров обнаруживают холестерол, окруженный липидными каплями (так наз. псевдоксантомные клетки). В мозге находят ангиомы, ангиоретикулемы, кистозные образования, различные аномалии желудочков, оболочек. При развитии ангиоретикулем с мультифокальным ростом отмечаются множественные кисты и полости, нередко возникают кровоизлияния. Обнаруживаются разнообразные конгенитальные аномалии развития внутренних органов: сердечно-сосудистой системы — коарктация аорты (см.), врожденные пороки сердца (см. Пороки сердца врожденные); поликистоз почек, поджелудочной железы, печени. Встречаются также феохромоцитомы (см.), гипернефромы (см. Надпочечники, опухоли).
Классической триадой Г.—Л. б. являются перечисленные выше сосудистые опухоли и аномалии развития сетчатки глаза, головного и спинного мозга, внутренних органов. Однако образование опухолевой ткани в организме обычно происходит неравномерно. Чаще встречаются неполные формы болезни, когда в одних случаях преобладает ангиоматоз сетчатки, в других на передний план выдвигается картина внутричерепной ангиоретикулемы (см.).
Клиническая картина
У членов одной семьи встречаются разнообразные сочетания симптомов Г.—Л.б. Она начинается в период от 10 до 30 лет. Ранними признаками часто бывают офтальмол. расстройства, проявляющиеся прогрессирующим снижением зрения и изменениями на глазном дне. Затем присоединяются симптомы поражения мозга. При ангиоматозе сетчатки наблюдается своеобразная картина глазного дна. В периферической части сетчатки, часто в нижнем сегменте, видны шаровидные красноватые возвышения. Они и представляют собой описанные выше капиллярные ангиомы. От диска зрительного нерва к ним подходит несколько крупных петлистых питающих сосудов. Ангиомы могут быть одиночными и множественными, развиваются на одном или обоих глазах. Оба глаза поражаются в 36—50%. В течении ангиоматоза сетчатки различают четыре стадии. В начальной стадии ангиомы развиваются в месте соустья нескольких расширенных артериальных и венозных ветвей, имеют малые размеры, ткань сетчатки относительно сохранена. Во второй стадии величина ангиом становится больше и они вдаются в стекловидное тело. Развивается реактивный глиоз, резкое расширение сосудов, экссудация, дистрофические изменения и кровоизлияния в сетчатке. В третьей стадии экссудация нарастает вплоть до образования сегментарной отслойки сетчатки. В четвертой — терминальной — стадии наступает тотальная отслойка сетчатки, резкая дистрофия всех структур глазного яблока.
Неврол, симптомы при Г.—Л. б. зависят от локализации ангиоретикулем в той или другой области мозга. Чаще всего эти опухоли развиваются в мозжечке, реже в продолговатом мозге, подкорковых ганглиях и в больших полушариях головного мозга. Изредка они встречаются в спинном мозге и нервных корешках. Начало неврол, расстройств при Г.—Л. б. чаще наблюдается в возрасте от 20 до 40 лет. При расположении ангиоретикулемы в полушарии мозжечка рано появляются общемозговые симптомы, в частности периодически усиливающаяся, особенно по утрам, головная боль, к-рая может стать постоянной. Обычно головная боль бывает разлитой, но может локализоваться в области затылка и отдавать в шею, спину, иногда локализуется в области лба. Головная боль часто сопровождается рвотой. Выявляются застойные диски зрительных нервов, иногда вынужденная поза головы. На этом фоне постепенно развивается расстройство статики и походки, формируется картина мозжечковой атаксии (см.), обычно двусторонней, но с преобладанием на стороне опухоли.
При ангиоретикулематозе продолговатого мозга ранними очаговыми признаками являются рвота, икота, дисфагия, сердечные и дыхательные расстройства, что связывают с вовлечением в процесс ядер IX и X пар черепных нервов. Позднее присоединяется атаксия, зависящая не только от поражения заднемедиальных участков мозжечка, но и от ядер задних канатиков. Болезнь развивается в течение ряда лет. Общемозговые симптомы появляются поздно.
При супратенториальной локализации ангиоретикулематоза первыми появляются общемозговые симптомы, но протекают они сравнительно мягко. Головные боли возникают приступами, напоминая мигрень. Наблюдаются эпилептические припадки, иногда кортикального типа. Течению этой формы Г.—-Л. б. особенно свойственны обострения (расстройства кровообращения в опухолевой ткани, проявляющиеся усилением общемозговых и очаговых симптомов) с последующими ремиссиями.
Ангиоретикулемы спинного мозга могут вызывать корешковые боли, выпадение сухожильных рефлексов и расстройства глубокой чувствительности (результат задней локализации опухоли в позвоночном канале). Иногда наступает картина поперечного спинального поражения. Встречается сочетание Г.—Л. б. с сирингомиелией (см.), сопровождающееся возникновением соответствующего симптомокомплекса. В цереброспинальной жидкости обнаруживают умеренную белково-клеточную диссоциацию; давление может быть повышено до 220, в отдельных случаях до 330 мм вод. ст. Ангиография (см.), пневмоэнцефалография (см. Энцефалография), сканирование (см.) способствуют установлению локализации, а иногда и типа опухоли.
Аномалии развития и новообразования внутренних органов при Г.—Л. б. развиваются скрыто и часто остаются нераспознанными. Развивающиеся из надпочечника гипернефромы и феохромоцитомы вызывают повышение АД.
Течение Гиппеля-Линдау болезни медленно прогрессирующее. Иногда наблюдаются ремиссии. Заболевание, начавшееся в детском возрасте, течет относительно благоприятно, в злокачественное может перейти в возрасте 35—40 лет и позже. При локализации ангиоретикулем в больших полушариях головного мозга, в мозжечке прогредиентность заболевания независимо от возраста чрезвычайно быстрая. Особенность течения Г.—Л. б. в детском возрасте — появление симптомов поражения нервной системы на фоне имеющихся офтальмологических изменений. В ряде случаев осложняется заболевание иридоциклитом (см.), вторичной глаукомой (см.), гемофтальмом (см.), катарактой (см.).
Диагноз
Диагноз в ранних стадиях благодаря характерной офтальмоскопической картине в сочетании с неврол, и соматическими симптомами не представляет затруднений. В поздних стадиях при наличии значительной экссудации заболевание необходимо дифференцировать с ретинитом Коутса (см. Ретинопатия), иногда — ретинобластомой (см.), что может быть решено с помощью выявления симптомов сочетанного поражения глаз и нервной системы, а также с опухолями нервной системы и внутренних органов.
Лечение
Прогноз
Прогноз зависит от формы заболевания, в ряде случаев неблагоприятный. При отсутствии лечения процесс прогрессирует, приводя к гибели глазного яблока, разрыву ангиом, аневризм с последующим кровоизлиянием в головной мозг. Своевременное хирургическое вмешательство устраняет мозговые расстройства.
Библиография Архангельский В. Н. Морфологические основы офтальмоскопической диагностики, с. 88, М., 1960; Кацнельсон А. Б. Ангиоматоз сетчатки, Сов. вестн. офтальм., т. 6, № 4, с. 555, 1935, библиогр.; Котелянский Э. О. Внутриглазные опухоли, с. 107, М., 1974, библиогр.; Левкоева Э. Ф. Болезнь Гиппель — Линдау (глиоматоз сетчатки) в морфологическом освещении, в кн.: Патология сетчатой оболочки и зрительного нерва, под ред. К. В. Трутневой, с. 185, М., 1971; Меркулов И. И. Клиническая офтальмология, кн. 2, с. 101, Харьков, 1971; Шепкалова В. М., Хорасанян-Тада А. А. и Дислер О. Н. Внутриглазные опухоли, с. 211, М., 1965; Шмидт Е. В. Ангиоретик у лома головного мозга, с. 136, М., 1955; Gahlot D. К., Кhosia P. К. a. PrakashP. Retino-cerebral angiomatosis, East. Arch. Ophthal., v. 2, p. 113, 1974; Hippel E. Uber eine sehr seltene Erkrankung der Netzhaut, Albrecht v. Graefes Arch. Ophthal., Bd 59, S. 83, 1904; Lindau A. Studien tiber Kleinhimcysten, Bau, Pathogenese und Beziehungen zur Angiomatosis retinae, Kobenhavn, 1926; Neurologie, hrsg. v. J. Quandt u. H. Sommer, Bd 2, S. 595, 803, Lpz., 1974; Rho J.M. Von Hippel — Lindau’s disease, Canad. med. Ass. J., v. 101, p. 135, 1969; Trevor-Poper P. D. The eye and its disorders, p. 648, Oxford a. o., 1974.
Болезнь Гиппеля-Линдау ( Цереброретинальный ангиоматоз )
Болезнь Гиппеля-Линдау — аутосомно-доминантная генная патология, обуславливающая развитие в организме целого ряда полиморфных опухолей. Наиболее часто это ангиомы сетчатки, гемангиобластомы ЦНС, феохромоцитомы, новообразования почек и поджелудочной железы. Иногда проявлением заболевания выступает единичный опухолевый процесс. Диагноз верифицируется после неврологического и офтальмологического обследований, проведения КТ или МРТ головного мозга и позвоночника, УЗИ или КТ почек, поджелудочной железы, надпочечников, генетической диагностики. Лечение состоит в раннем выявлении и удалении появляющихся опухолевых образований.
МКБ-10

Общие сведения
Болезнь встречается с частотой 1 случай на 36 тыс. чел. Отличается большим полиморфизмом и различной локализацией возникающих опухолей. Наиболее распространенным признаком является ретинальный ангиоматоз, который сопровождает до 75% случаев заболевания. Зачастую он выступает диагностическим маркером данной патологии. Гемангиобластомы мозжечка по различным данным наблюдаются в 35-70% случаев, новообразования и кисты почек — у 25% больных, поражение поджелудочной железы — у 24%, феохромоцитома — у 7%. По причине большой вариабельности новообразований пациенты, имеющие болезнь Гиппеля-Линдау, нуждаются в совместной курации специалистов в области офтальмологии, неврологии, онкологии, урологии, гастроэнтерологии, эндокринологии.
Причины болезни Гиппеля-Линдау
Болезнь Гиппеля-Линдау является генной патологией. Примерно в 80% случаев она наследуется аутосомно-доминантным способом с неполной пенетрантностью гена. Еще в 20% случаев болезнь Гиппеля-Линдау возникает вследствие новых мутаций. Аберрации затрагивают расположенный в 3-ей хромосоме участок р25-26, а именно ген VHL, который играет роль супрессора, подавляющего рост новообразований. На сегодняшний день известно около 140 мутаций данного гена.
В результате недостаточной онкосупрессии происходит рост новообразований, преимущественно ангиоретикулом и гемангиобластом. Опухоли поражают мозжечок и сетчатку глаза, реже отмечаются внутримозговые опухоли полушарий, новообразования подкорковых структур и продолговатого мозга, еще реже — опухоли спинного мозга и периферических нервов. Из-за неполной проявленности генетических аберраций у некоторых пациентов может наблюдаться лишь один клинический признак болезни.
В соответствии с классификацией, болезнь Гиппеля-Линдау имеет 2 типа: без феохромоцитомы и с ее наличием. Второй тип подразделяется на варианты: 2А — с низким риском развития аденокарциномы почки, 2В — с высоким риском карциномы, 2С — наблюдается только феохромоцитома. При всех вариантах заболевания, кроме 2С, возможно наличие гемангиобластом ЦНС и ангиом сетчатки.
Симптомы болезни Гиппеля-Линдау
Дебют неврологических проявлений обычно приходится на 3-4-е десятилетия жизни. В детском возрасте болезнь Гиппеля-Линдау отличается появлением неврологической симптоматики на фоне уже существующих зрительных расстройств. В ряде случаев заболевание у детей манифестирует субарахноидальным кровоизлиянием.
Поражение ЦНС. Наиболее часто источником первичных симптомов выступают церебеллярные кисты (кисты мозжечка). Они манифестируют общемозговыми симптомами (диффузными головными болями, тошнотой без связи с приемом пищи, рвотой, шумом в ушах), обусловленными повышением внутричерепного давления. К первым признакам также относятся эпиприступы, они могут быть генерализованными либо фокальными. Со временем проявляются признаки поражения мозжечка, формирующие симптомокомплекс мозжечковой атаксии: статическая и динамическая дискоординация, адиадохокинез, гиперметрия и асинергия, интенционный тремор, миодистония. По мере роста церебеллярного новообразования возникает смещение и сдавление мозгового ствола, сопровождающееся стволовыми симптомами, в первую очередь, расстройством глотания, диплопией, дизартрией. Спинальные опухоли (чаще ангиоретикуломы) проявляются корешковыми синдромами, выпадением глубоких видов чувствительности, отсутствием сухожильных рефлексов. В 80% случаев спинальной патологии отмечается клиника, сходная с сирингомиелией. Возможна картина полного поражения поперечника спинного мозга.
Поражение глаз на ранних стадиях диагностируются лишь при офтальмоскопии. После 8 лет появляются жалобы на туманность изображения и его искажение (метаморфопсии). У половины пациентов выявляется поражение обоих глаз. Увеличивающиеся со временем ангиомы сетчатки приводят к расстройству кровообращения в ее сосудах, ишемии и кистозной дегенерации. В поздней стадии возможны увеит, катаракта, отслойка сетчатки, глаукома, гемофтальм.
Поражение почек в 60-90% случаев представлено кистами, в 45% случаев — ренальноклеточной карциномой. Как правило, почечная карцинома клинически дебютирует в возрасте от 40 до 50 лет у больных, которые ранее уже лечились по поводу новообразований. В половине случаев на момент диагностирования карциномы выявляются ее метастазы. Сочетание поликистоза почек с ангиоматозом сетчатки более характерно, чем его комбинация с церебральными ангиомами. У 35% пациентов, имеющих болезнь Гиппеля-Линдау, поликистоз диагностируется посмертно. В детском возрасте при семейном типе заболевания поликистоз почек зачастую является его единственным проявлением.
Феохромоцитома почти в половине случаев имеет двусторонний характер. Может выступать единственным клиническим проявлением болезни. В сочетании с почечной карциномой наблюдается довольно редко.
Поражение поджелудочной железы от 30 до 72% составляют ее кисты. Кисты поджелудочной железы носят доброкачественный характер и редко приводят к клинически значимой ферментативной недостаточности панкреас. Хотя известны случаи полного замещения кистой нормальных тканей железы с развитием сахарного диабета.
Диагностика болезни Гиппеля-Линдау
Полная верификация диагноза осуществляется коллегиально неврологом, офтальмологом и генетиком при участии других врачей: онколога, эндокринолога, уролога, гастроэнтеролога.
На начальном этапе проводят полный неврологический и офтальмологический осмотр. С целью выявления церебеллярных образований назначают КТ или МРТ головного мозга. Для обнаружения опухолей другой локализации необходимо УЗИ или КТ почек, УЗИ поджелудочной железы или ее МРТ, МРТ позвоночника, КТ надпочечников. Проводится анализ уровня катехоламинов и ферментов поджелудочной железы. ДНК-диагностика направлена на выявление мутаций в VHL-гене.
Предполагать и исключать болезнь Гиппеля-Линдау следует в каждом случае выявления ангиоматоза сетчатки в ходе офтальмоскопии, особенно при наличии отягощенного семейного анамнеза. В начальной стадии офтальмоскопия может определять одиночную ангиому сетчатки с дилатацией питающих ее сосудов, впоследствии ангиомы становятся множественными, характерны аневризмы и змееобразная извитость сосудов. Диагностировать самые ранние изменения сосудов сетчатки и стертые формы позволяет флюоресцентная ангиография сетчатки. С ее помощью можно дифференцировать изменения сетчатки, сопровождающие болезнь Гиппеля-Линдау, от другой офтальмологической патологии: ретинопатий, ритинита, ретинобластомы, нейропатии зрительного нерва и пр. Уточнение диагноза возможно при помощи лазерной томографии сетчатки.
Лечение и прогноз болезни Гиппеля-Линдау
Сегодня болезнь Гиппеля-Линдау имеет лишь симптоматическое лечение. Оно направлено на ликвидацию возникающих опухолевых образований. Для как можно более раннего выявления опухолей рекомендовано наблюдение и ежегодное обследование пациентов.
Ранние стадии ангиоматоза сетчатки являются показанием к фокусной лучевой терапии, однако через год после ее проведения может возникнуть радиационная ретинопатия. В отношении ангиом небольшого размера возможна лазерная коагуляция, диатермокоагуляция, при больших образованиях — транссклеральная криопексия. Если болезнь Гиппеля-Линдау сопровождается новообразованиями ЦНС, необходима консультация нейрохирурга.
Возможно хирургическое удаление опухоли мозжечка, полушарий мозга, зрительного нерва. Описаны случаи применения стереотаксической хирургии. При диагностировании почечной карциномы производится частичная нефрэктомия, при выявлении феохромоцитомы — ее удаление. Хирургическое лечение доброкачественных новообразований поджелудочной железы показано при увеличении их размеров свыше 2-3 см.
Без проведения лечения заболевание приводит к слепоте вследствие прогрессирующего ангиоматоза сетчатки и к летальному исходу вследствие развития опухолей церебральной и соматической локализации. При наблюдении и лечении пациенты доживают в среднем до 40-50-летнего возраста. Половина летальных исходов обусловлена гемангиобластомами ЦНС. На ранних стадиях радикальное удаление этих опухолей удается у большинства больных, однако новообразования склонны рецидивировать в среднем через 6 лет после их удаления.
Болезнь Гиппеля-Линдау
Болезнь Гиппеля-Линдау (или синдром фон Гиппеля-Линдау, англ. Von Hippel-Lindau disease — VHL) — это аутосомно-доминантное генетическое заболевание, возникающее в результате делеции или мутации в гене VHL. Синдром Гиппеля-Линдау поражает 1 из 36 000 человек (200 000 случаев во всем мире), и 20% пациентов являются первыми в семье (т.е новая мутация). Средний возраст начала заболевания составляет 26 лет, и 97% людей с мутацией гена VHL имеют симптомы к 65 годам. Болезнь Гиппеля-Линдау поражает мужчин и женщин и все этнические группы в равной степени и встречается во всех частях мира. У пациентов с заболеванием могут возникать опухоли и/или кисты в десяти частях тела, включая мозг, позвоночник, глаза, почки, поджелудочную железу, надпочечники, внутреннее ухо, репродуктивные пути, печень и легкие с последующими симптомами/осложнениями:
- Гемангиобластома головного мозга/позвоночника: головные боли, атаксия, нистагм, боль в спине, онемение, икота;
- Гемангиобластома сетчатки:плавающие помутнения — «мушки» в глазах, отслоение сетчатки;
- Опухоли эндолимфатического мешка: потеря слуха, тиннитус (звон/шум в ушах), головокружение;
- Кисты/опухоли/рак поджелудочной железы:панкреатит (из-за закупорки желчных протоков), диабет (из-за блокировки доставки инсулина), раздражительность пищеварения, мальабсорбция кишечника, желтуха;
- Феохромоцитома, параганглиома: высокое кровяное давление (артериальная гипертензия), панические атаки (или послеоперационная надпочечниковая недостаточность);
- Кисты почек, почечно-клеточная карцинома: боль в пояснице, гематурия, утомляемость;
- Цистаденома (у мужчин и женщин): боль, разрыв, кровотечения, перекрут (возможен рак яичников)
Большинство этих опухолей доброкачественные, но это не означает, что они безобидные. На самом деле доброкачественные опухоли синдрома Гиппеля-Линдау все еще могут быть очень серьезными. По мере увеличения размера эти опухоли и связанные с ними кисты могут оказывать повышенное давление на структуру вокруг них. Это давление может вызвать симптомы, включая сильную боль или еще более серьезные осложнения.
Болезнь Гиппеля-Линдау отличается у каждого пациента, даже в пределах одной семьи. Поскольку невозможно точно предсказать, как и когда заболевание проявится у каждого человека, очень важно регулярно проверять возможные проявления расстройства на протяжении всей жизни человека.
В настоящее время не существует лекарственного (фармакологического) лечения; хирургическое удаление — основной метод лечения. Подход с сохранением органов — лучший подход для уменьшения непоправимого ущерба при минимальном удалении органов. При тщательном мониторинге, раннем обнаружении и соответствующем лечении наиболее вредные последствия мутации VHL гена могут быть значительно уменьшены, а у некоторых людей полностью предотвращены.
Поскольку заболевание может вызывать злокачественные опухоли, оно считается одним из группы факторов риска семейного рака, которые передаются генетически. Цель состоит в том, чтобы найти опухоль на ранней стадии, следить за признаками роста опухоли и удалить или вылечить опухоль до того, как она проникнет в другие ткани. Доброкачественные опухоли также могут нуждаться в лечении или удалении, если их рост вызывает симптомы.
Признаки и симптомы
Болезнь Гиппеля-Линдау не имеет ни одного первичного симптома. Частично это связано с тем, что он возникает не только в одном органе тела. Синдром также не всегда возникает в определенной возрастной группе. Это наследственное заболевание, но его проявления могут сильно отличаться у разных людей, несмотря на одну и ту же генетическую мутацию. Кроме того, внешний вид и тяжесть поражений у разных людей настолько различаются, что многие члены одной семьи могут иметь только некоторые относительно безобидные проблемы, в то время как другие могут иметь серьезные осложнения.
Возраст начала варьируется от семьи к семье и от человека к человеку. Феохромоцитома (рак надпочечников) очень распространена в некоторых семьях, тогда как светлоклеточный почечно-клеточный рак ([опухоль почек]) более распространен в других семьях.
Наиболее частый симптом синдрома Гиппеля-Линдау — гемангиобластомы. Это доброкачественные опухоли головного, спинного мозга и сетчатки. Гемангиобластомы доброкачественные. В головном или спинном мозге гемангиобластома в некоторых случаях может находиться внутри кисты или мешка, заполненного жидкостью. Гемангиобластомы или окружающие кисты могут давить на нерв или ткань мозга и вызывать такие симптомы, как:
- головные боли;
- проблемы с равновесием при ходьбе;
- слабость рук и ног.
Кровоизлияние в глаза или утечка жидкости из гемангиобластом может мешать зрению. Раннее обнаружение, тщательное наблюдение за глазами и своевременное лечение очень важны для поддержания здорового зрения.
Ранними признаками опухолей надпочечников могут быть высокое кровяное давление, панические атаки или сильное потоотделение. Ранние признаки кист и опухолей поджелудочной железы могут включать жалобы на пищеварение, такие как вздутие живота или нарушение функции кишечника и мочевого пузыря. Некоторые из этих опухолей доброкачественные, а другие могут стать злокачественными.
Опухоли и кисты почек (светлоклеточная почечно-клеточная карцинома) могут приводить к снижению функции почек, но обычно на ранних стадиях симптомы отсутствуют. Если опухоль почек не будет удалена, то метастазы начнут распространятся, когда они достигнут примерно 3 см в диаметре.
Болезнь Гиппеля-Линдау может также вызвать доброкачественную опухоль во внутреннем ухе, называемую опухолью эндолимфатического мешка. Если эту опухоль не удалить, она может привести к потере слуха в пораженном ухе, а также к нарушению равновесия. Менее распространенные проявления синдрома включают доброкачественные опухоли половых путей как у мужчин, так и у женщин. Однако эти опухоли могут привести к проблемам с оплодотворением или беременностью.
Опухоли в печени и легких считаются безобидными.
Причины
Болезнь Гиппеля-Линдау — это аутосомно-доминантное заболевание, возникающее в результате делеции или мутации гена VHL, расположенного на коротком плече хромосомы 3. Каждый ребенок человека с синдромом подвергается 50% риску унаследовать измененную копию гена VHL.
Нормальный ген VHL действует как ген-супрессор опухолей с функцией предотвращения образования опухолей. Ген действует как ключевой регулятор передачи сигналов клеточной гипоксии через свой продукт, белок VHL (pVHL). pVHL через комплекс HIF (фактор, индуцируемый гипоксией) косвенно отвечает за повышенные уровни факторов роста, включая фактор эндотелия сосудов, фактор роста, полученный из тромбоцитов, и трансформирующий фактор роста альфа.
В случае нефункционирующего гена, например, при болезни Гиппеля-Линдау, регуляция комплекса HIF не происходит. В результате повышается уровень различных факторов роста, что способствует усилению роста кровеносных сосудов (ангиогенезу) и образованию опухолей. Это тот же процесс, что и при других более распространенных формах рака, таких как рак почки, груди, поджелудочной железы, надпочечников.
Исследователи считают, что вмешательство в активность фактора роста и/или HIF окажется эффективным лечением синдрома Гиппеля-Линдау и других форм рака.
Затронутые группы населения
Болезнь является наследственным генетическим заболеванием, однако 20% всех пациентов являются первыми в семье. Заболеваемость составляет 1 из 36 000 рождений, поражая в равной степени мужчин и женщин, а также все этнические группы, и происходит во всех частях мира.
Связанные расстройства
Болезнь Гиппеля-Линдау — сложное заболевание, которое вызывает рост опухолей в 10 различных частях тела: почках, надпочечниках, поджелудочной железе, головном мозге, позвоночнике, сетчатке, внутреннем ухе, репродуктивном тракте, печени и легких. Из-за поражения многих органов симптомы и проявления синдрома совпадают с широким спектром заболеваний. К ним относятся следующие:
- Почки: спорадический рак почки, синдром Бёрта-Хога-Дьюба, наследственный лейомиоматоз и почечно-клеточная карцинома, комплекс туберозного склероза, избыток сукцинатдегидрогеназы.
- Надпочечник или феохромоцитома: избыток сукцинатдегидрогеназы, множественная эндокринная неоплазия 2 типа, типы A и B (МЭН2A и МЭН2B).
- Внутреннее ухо:болезнь Меньера.
- Поджелудочная железа: рак поджелудочной железы.
- Сетчатка: гемангиобластомы сетчатки уникальны для болезни Гиппеля-Линдау. Наличие гемангиобластомы сетчатки приводит к клиническому диагнозу заболевания.
- Мозг или позвоночник: гемангиобластомы в головном или спинном мозге отличаются от других форм опухолей головного или спинного мозга, и поэтому их диагноз считается критерием для генетического анализп мутации гена VHL. Обратите внимание, что исследования других типов опухолей головного мозга не имеют отношения к гемангиобластомам болезни Гиппеля-Линдау.
Диагностика
Любой, у кого есть родитель с синдромом Гиппеля-Линдау, и большинство людей, у которых есть брат или сестра с болезнью, имеют 50%-ную вероятность заболевания. Любой человек, у которого есть тетя, дядя, двоюродный брат или дедушка или бабушка с заболеванием, также может подвергаться риску. Единственный способ точно определить, что у кого-то нет мутации гена VHL — это ДНК тест. Клинический диагноз также может быть поставлен, когда у человека обнаруживается опухоль, специфичная для заболевания.
После того, как диагноз болезни Гиппеля-Линдау поставлен, важно начать контрольное обследование как можно раньше, до появления каких-либо симптомов. Большинство опухолей/кист гораздо легче лечить, когда они маленькие. Ряд возможных осложнений болезни Гиппеля-Линдау не проявляются симптомами, пока проблема не разовьется до критического уровня. Лечение может только остановить появившиеся симптомы; не всегда возможно отменить изменения и вернуться к нормальному состоянию.
Стандартные методы лечения
Универсальной рекомендации по лечению не существует. Варианты лечения могут быть определены только путем тщательной оценки общей ситуации отдельного пациента — симптомов, результатов тестов, визуализационных исследований и общего физического состояния. В качестве общих рекомендаций по возможному способу лечения предлагаются следующие.
— Гемангиобластомы головного и спинного мозга.
Симптомы, связанные с гемангиобластомами головного и спинного мозга, зависят от расположения, размера опухоли и наличия связанного с ней отека или кисты. Симптоматические поражения растут быстрее, чем бессимптомные. Кисты часто вызывают больше симптомов, чем сама опухоль. Как только поражение будет удалено, киста разрушится. Если какая-либо часть опухоли останется на месте, киста снова заполнится. Небольшие гемангиобластомы, которые не имеют симптомов и не связаны с кистой, иногда лечат стереотаксической радиохирургией, но это больше профилактика, чем лечение, и отдаленные результаты, кажется, показывают лишь незначительную пользу. Кроме того, в период восстановления симптомы могут не уменьшиться.
— Нейроэндокринные опухоли поджелудочной железы.
Для дифференциации серозных цистаденом от нейроэндокринных опухолей поджелудочной железы (НЭО поджелудочной железы) необходим тщательный анализ. Кисты и цистаденомы обычно не требуют лечения. НЭО поджелудочной железы следует оценивать по размеру, поведению и конкретной генетической мутации.
— Почечно-клеточная карцинома.
Опухоли почек болезни Гиппеля-Линдау часто обнаруживаются, когда они очень маленькие по размеру и находятся на очень ранних стадиях развития. Стратегия обеспечения того, чтобы у человека была достаточно функционирующая почка на протяжении всей его жизни, начинается с тщательного наблюдения и выбора операции только тогда, когда размер опухоли или высокая скорость роста предполагают, что опухоль может получить метастатический потенциал (примерно на 3 см). При этом широко используется методика орган-сохраняющей хирургии. Радиочастотная абляция (РЧА) или криохирургия (криотерапия) могут быть рассмотрены, особенно для небольших опухолей на ранних стадиях. Необходимо соблюдать осторожность, чтобы не повредить соседние структуры и ограничить образование рубцов, которые могут осложнить последующие операции.
— Гемангиобластомы сетчатки.
Небольшие периферические поражения можно успешно лечить с минимальной потерей зрения или без нее с помощью лазера. Большие поражения часто требуют криотерапии. Если гемангиобластома находится на диске зрительного нерва, существует несколько вариантов лечения, которые позволят успешно сохранить зрение.
— Феохромоцитомы.
Хирургическое удаление проводится после адекватной блокады медикаментами, предпочтительна лапароскопическая частичная адреналэктомия. Жизненно важные показатели тщательно контролируются в течение как минимум недели после операции, пока организм приспосабливается к своей «новой норме». Особая осторожность требуется во время хирургических вмешательств любого типа, а также во время беременности и родов. Даже феохромоцитомы, которые не выглядят активными или не вызывают симптомов, следует рассматривать для удаления, в идеале до беременности или неэкстренного хирургического вмешательства.
— Опухоли эндолимфатического мешка.
Пациентам, у которых опухоль или кровоизлияние видно на МРТ, но которые все еще слышат, требуется хирургическое вмешательство, чтобы предотвратить ухудшение их состояния. Глухие пациенты с данными визуализации опухоли должны подвергнуться хирургическому вмешательству, если присутствуют другие неврологические симптомы, чтобы предотвратить ухудшение баланса. Не все опухоли эндолимфатического мешка видны при визуализации; некоторые обнаруживаются только во время операции.
Прогноз болезни Гиппеля-Линдау зависит от возникновения множественных опухолей. Почечно-клеточная карцинома является основной причиной смерти, за ним следуют гемангиобластомы центральной нервной системы. Средняя продолжительность жизни ранее оценивалась в 50 лет; однако регулярное наблюдение, раннее выявление и лечение опухолей в настоящее время снизили заболеваемость и смертность.

Врач-терапевт, гастроэнтеролог, гепатолог, инфекционист. Провожу профилактические мероприятия осложнений со стороны пищеварительной системы после долгой терапии НПВП и кроворазжижающими препаратами.
Болезнь Гиппеля-Линдау — когда опухоли съедают организм

Болезнь Гиппеля-Линдау относится к категории сложных наследственных заболеваний.
Для этого нарушения характерно образование множественных опухолей различных органов.
Чтобы минимизировать негативное влияние данного недуга на организм человека, очень важно своевременно обратиться к врачу.
Благодаря ранней диагностике и своевременному лечению можно контролировать течение болезни.
Описание болезни
Под данным термином понимают нарушение наследственного характера, которое сопровождается чрезмерным ростом капилляров и появлением большого количества опухолей.
- гемангиобластомы - опухолевые образования нервной системы, которые локализуются в сетчатке глаз и мозжечке;
- ангиомы - сосудистые образования в спинном мозге;
- кисты - полостные образования, которые внутри наполнены жидкостью, они могут формироваться в почках, печени, яичках, поджелудочной железе;
- феохромоцитомы - опухолевые образования, состоящие из мозгового слоя надпочечников.
Механизм развития болезни
Синдром Гиппеля Линдау передается по аутосомно-доминантному типу. Это значит, что если один из родителей обладает дефектным геном, риск развития болезни у ребенка составляет 50 %.
Этот синдром появляется у людей разного возраста и имеет различные проявления даже у ближайших родственников.
Примерно 90 % людей с этим заболеванием к 65 годам обнаруживают у себя минимум один симптомов.
Пока не были разработаны методы ранней диагностики, пациенты обычно не доживали до пятидесятилетнего возраста.
В последние годы прогноз болезни заметно улучшился, поскольку существуют способы раннего выявления патологии и новые технологии ее лечения.
Характерные симптомы
Для болезни Гиппеля Линдау характерно достаточно много симптомов.
Так, нарушения в работе мозжечка имеют такие проявления:
- шаткость походки - во время ходьбы человек может раскачиваться в разные стороны;
- нарушение координации движений - человек промахивается при выполнении точных движений;
- тремор - наблюдается дрожание конечностей;
- нистагм - развиваются ритмичные колебания глазных яблок в стороны, причем для них характерна достаточнобольшая амплитуда размаха.
Также для этого заболевания характерно повышение внутричерепного давления, которое связано с нарушением оттока ликвора из мозга.
Это состояние характеризуется следующими проявлениями:
- головная боль, которая имеет разлитой характер;
- тошнота и рвота;
- повышенная утомляемость и слабость.
Заболевание сопровождается развитием опухолевых образований в различных органах и системах. Этот процесс обычно сопровождается следующими проявлениями:
- учащение сердцебиения и приступы жара в теле - характерны для опухолевого образования в мозговом слое надпочечника;
- отечность рук, ног, лица, появление крови в моче - наблюдается при патологических процессах в почках;
- бесплодие и болевые ощущения в яичках - характерны для опухолей яичек;
- диарея, ощущение сухости в ротовой полости и постоянная жажда - наблюдается при опухоли поджелудочной железы;
- снижение остроты зрения или полная его потеря - сопровождает появление сосудистой опухоли сетчатки.
Процесс развития — стремительно и необратимо
Существует несколько стадий развития данного заболеваний:
- Доклиническая. Для нее характерны начальные скопления капилляров и небольшое их расширение.
- Классическая. На этом этапе формируются типичные ангиомы сетчатки.
- Экссудативная. Для нее характерна высокая проницаемость стенок ангиоматозных узлов.
- На четвертом этапе происходит отслойка сетчатки, причем она может иметь тракционный или экссудативный характер.
- Терминальная. На этой стадии развивается глаукома, увеит, фтизис глазного яблока.
Если начать лечение болезни Гиппеле Линдау на раннем этапе, можно добиться достаточно эффективных результатов. Кроме того, благодаря своевременной терапии удается минимизировать угрозу развития осложнений.
Для болезни Гиппеля-Линдау является характерным интенционный тремор. Какие заболевания еще сопровождает этот симптом.
Астроцитома головного мозга — серьезная опухоль, которая может привести к летальному исходу. Какие методы удаления образования существуют?
Чтобы поставить правильный диагноз, врач должен проанализировать жалобы пациента и изучить историю болезни.
Специалиста будет интересовать такая информация:
- когда возникли головные боли, ухудшение остроты зрения, шаткость походки;
- имеются ли у ближайших родственников аналогичные симптомы;
- есть ли у членов семьи онкологические заболевания.
Затем проводится неврологический осмотр, во время которого нужно оценить работу мозжечка. Для этого определяют наличие шаткости походки и проблем с координацией движений.
Следующим этапом диагностики является осмотр глазного дна, который позволит выявить опухолевые образования сетчатки и симптомы внутричерепного давления - они заключаются в бледности и размытости границ диска зрительного нерва.
Большую диагностическую ценность имеет также анализ крови. При развитии опухоли в поджелудочной железе данное исследование отобразит увеличение объема глюкозы в крови.
Если у человека наблюдается феохромоцитома, в крови повышается уровень катехоламинов - именно эти вещества синтезируются мозговым веществом.
Помимо этого, диагностика болезни должна включать такие исследования:
- Компьютерная и магнитно-резонансная томография. При помощи данных процедур можно оценить строение мозга и выявить опухолевые образования в мозжечке. Помимо этого, удается обнаружить симптомы повышения внутричерепного давления.
- Ультразвуковое исследование. Обычно исследуют органы, которые могут быть поражены данным недугом, - почки, печень, поджелудочную железу, яички.
- Исследование генеалогического дерева. Данная процедура заключается в подробной беседе с пациентом и его родственниками. Врач должен получить информацию относительно наличия таких жалоб у близких родственников.
- Консультация узких специалистов. При подозрении на болезнь Гиппеля-Линдау человека направляют к генетику, офтальмологу, нейрохирургу.
Методы лечения
Пока опухолевые образования имеют небольшие размеры и не связаны ни с какими симптомами, их не удаляют.
Иссечение гемангиобластомы проводится в том случае, если опухоль имеет четкие признаки и является операбельной.
При размерах образования менее 3 см может применяться лучевая терапия. Если опухоль локализуется в сетчатке глаза, нужно немедленно проводить лазерную коагуляцию или криотерапию.
При хирургическом лечении болезни нужно учитывать, что опухоли обычно имеют множественный характер. В любое время могут появиться новые образования, поэтому полностью вылечить пациента оперативным путем невозможно. Осторожно, видео операции! Кликните, чтобы открыть
Пациенты с этим заболеванием должны находиться под постоянным врачебным контролем, который заключается в проведении магнитно-резонансной томографии головы и спинного мозга.
На основании результатов этого исследования врач принимает решение относительно необходимости удаления конкретных опухолей.
В качестве дополнительных средств могут использоваться лекарственные препараты.
При болезни Гиппеля-Линдау часто применяют ноотропы - средства для улучшения работы головного мозга. При повышении объема глюкозы в крови показано использование сахароснижающих препаратов.
Осложнений не избежать
Это достаточно серьезное нарушение может спровоцировать такие последствия:
- полная потеря зрения - связана с увеличением размеров опухоли и отслоением сетчатки;
- смерть - при сдавливании жизненно важных органов может произойти остановка дыхания и кровообращения.
Прогноз при этом заболевании является достаточно неблагоприятным. Разрыв ангиом и аневризм может спровоцировать кровоизлияние в мозг и другие органы, что приведет к летальному исходу.
В среднем продолжительность жизни с цереброретинальным антиоматозом составляет 40±9 лет. Самой частой причиной смерти при этом заболевании является церебеллярная гемангиобластома.
Профилактики нет — помогут только молитвы
Профилактика данного заболевания невозможна, поскольку оно носит наследственный характер.
Людям, которые страдают этим недугом и планируют рождение ребенка, нужно обратиться к генетику. Благодаря специальному обследованию удастся оценить вероятность развития этой болезни у ребенка.
Болезнь Гиппеля-Линдау - крайне серьезное нарушение наследственного характера, которое сопровождается образованием множественных опухолей.
Чтобы избежать опасных последствий для здоровья, очень важно обратиться к врачу на раннем этапе развития болезни и постоянно держать течение патологии под контролем.
Синдром Линдау (Lindau) - синонимы, авторы, клиника
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Болезнь фон Гиппеля-Линдау (VHL-синдром)
Болезнь фон Гиппеля-Линдау является наследственным опухолевым синдромом, предполагающим развитие различных доброкачественных и злокачественных новообразований (гемангиобластома центральной нервной системы и сетчатки глаза, опухоль внутреннего уха, карцинома и кисты почек, феохромоцитома, нейроэндокринная опухоль и кисты поджелудочной железы, цистаденома придатка яичка у мужчин и широкой связки у женщин). Болезнь фон Гиппеля-Линдау - наиболее распространенная причина наследственного рака почки.
В 1895 г. Ю. фон Гиппель [1] описал пациента с ретинальной ангиомой, а в 1926 г. А. Линдау [2] — пациента с ретинальной ангиомой и гемангиоматозом центральной нервной системы. Год спустя тот же автор обнаружил ассоциацию этих проявлений с почечными и панкреатическими кистами [3]. Термин «синдром von Hippel—Lindau» (VHL) был введен Мелмоном и Роузеном [4]. Этот синдром выявляется приблизительно у 1 из 36 000 человек [5] и обусловлен мутацией в участке 3p25/26, где локализован ген подавления роста опухоли VHL [6—8]. 23% пациентов не имеют семейного анамнеза заболевания [9—13].

Ген VHL был идентифицирован в 1993 г. [8, 14]. Приблизительно у 20% пациентов выявляется делеция VHL-локуса в материнской или отцовской аллели [15, 16]. Герминальные мутации VHL наследуются по аутосомно-доминантному типу. Почти все мутации VHL у пациентов с феохромоцитомой являются миссенс-мутациями. Ген VHL состоит из 3 экзонов. Белок VHL (pVHL) включает 213 аминокислотных остатков, и его молекулярная масса равна приблизительно 28 кДa. Клетки с дефицитом pVHL накапливают фактор, индуцирующий гипоксию (HIF), что приводит к перепроизводству HIF-зависимых продуктов (которые вовлечены в адаптацию к гипоксии): сосудистого эндотелиального фактора роста (VEGF), эритропоэтина и трансформирующего ростового фактора альфа (TGF). Это объясняет сильную васкуляризацию VHL-ассоциированных опухолей [17—19]. Таким образом, продукт мутированного гена VHL приводит к сверхрегулированию различных генов, участвующих в патогенезе гипоксии, ускоряет ангиогенез, изменяет внеклеточный матрикс и регуляцию клеточного цикла [20—28]. Однако точные механизмы туморогенеза при синдроме VHL в настоящее время остаются неизвестными (рис. 1). Рисунок 1. Комплекс VBC (VHL protein and Elongin B, C) и механизм его действия. а — Normoxic condition — нормальное давление кислорода; Ub — убиквитин; HIF — фактор, индуцирующий гипоксию; pVHL — VHL-протеин; α — элонгин C-связывающий домен pVHL; β — β-домен (субстратсвязывающий домен) pVHL; CUL2 — куллин-2, формирующий комплекс с элонгином-B, C и pVHL; б — мутация гена VHL и, как результат, отсутствие регуляции HIF, VEGF, PDGF и TGF-α; в — мутация гена VHL и отсутствие регуляции aPKC λ (атипичная протеинкиназа C) [29].
VHL-синдром характеризуется развитием гемангиобластом сетчатки глаза (ангиомы сетчатки) и центральной нервной системы (ЦНС), билатеральной и мультифокальной дифференцированной карциномы почки, поликистоза почек, феохромоцитомы, кист и нейроэндокринных опухолей поджелудочной железы, папиллярной цистаденомы придатка яичка у мужчин и широкой связки у женщин, опухолей внутреннего уха. Поражение различных органов и степень этого поражения очень вариабельны (табл. 1, 2).
Клинически заболевание делится на две группы. Тип 1 включает главным образом большие делеции или мутации гена VHL и характеризуется полным фенотипом заболевания [поражение сетчатки, кисты или опухоли головного и спинного мозга, панкреатические, почечные, и селезеночные кисты, солидные панкреатические опухоли (реже аденокарциномы), карциномы почек, цистаденомы эпидидимуса и опухоль внутреннего уха], но без феохромоцитомы. Тип 2, при котором развивается феохромоцитома (миссенс-мутации гена VHL может иметь и неполный фенотип [13, 37, 40]. Тип 2 подразделяется на подтипы с низким (тип 2A) и высоким (тип 2B) риском развития рака почки, а также тип 2C, проявляющийся только феохромоцитомой [35, 38, 41—43] (табл. 3).

Гемангиобластомы ЦНС могут выявляться в детском возрасте, однако средний возраст диагностирования составляет 29 лет [32, 45, 46]. VHL-ассоциированные гемангиобластомы выявляются в среднем на 15 лет раньше, чем спорадические [47]. В зависимости от размера и местоположения опухоли клинические признаки гемангиобластомы ЦНС включают головную боль, тошноту, головокружение, атаксию, расстройство координации движений, нистагм, расстройства речи. Гемангиобластома спинного мозга может приводить к слабости конечностей и парестезиям. Диагноз устанавливается с помощью МРТ головного мозга и позвоночника. Гемангиобластомы обычно характеризуются медленным ростом и имеют высокий риск кровотечений, часто являются мультифокальными. Понимание патогенеза заболевания важно для выбора оптимального времени скрининга на опухоли и лечение [19]. Исследование тканей ЦНС умерших пациентов помогло пониманию гистогенеза гемангиобластом [49]. Активация фактора, индуцирующего гипоксию 2-альфа (HIF 2-α) происходит в маленьких мезенхимальных опухолях и в мезенхимальном компоненте больших опухолей. Активация HIF 1-α наблюдается в эпителиальном компоненте. Это позволило предполагать, что поражение ЦНС при VHL-синдроме — длительный процесс гемангиобластической пролиферации и дифференцировки [50] (рис. 2). Рисунок 2. Гемангиобластома ЦНС.
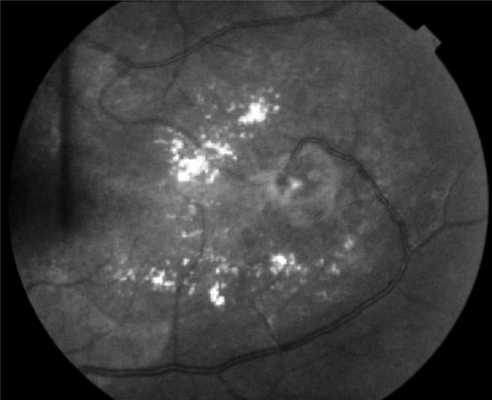
Поражения глаз выявляются примерно у 37% пациентов с VHL-синдромом, среди них только у 14% обнаруживается полная делеция VHL [51, 52]. Приблизительно у 8% пациентов снижена острота зрения [53]. Для лечения ангиомы сетчатки используют лазерную или криотерапию [32, 34, 54]. Недавние исследования [ 55, 56] показали, что при внутривенном введении антагониста сосудистого эндотелиального фактора роста (anti-VEGF) в течение 7 мес размер гемангиобластом не уменьшается (рис. 3). Рисунок 3. Ангиоматоз сетчатки.
У пациентов с синдромом VHL могут встречаться как кисты, так и рак почек [57—62]. Средний возраст манифестации — 37 лет. Для диагностики используют КТ и УЗИ [36, 63, 64]. Поскольку со`лидные раки могут содержать кистозные части (что затрудняет дифференцирование доброкачественных и злокачественных процессов с помощью визуализирующих методик), при отсутствии данных о метастазах лечение должно быть направлено на удаление этих образований по возможности с соблюдением принципа органосохраняющей операции. Это позволяет поддерживать почечную функцию максимально долго и избежать диализа [58, 65]. Опухоли почек отличаются медленным ростом (3 см (по стандартам США) или 5 см (по стандартам Европы) [58, 60, 67, 68]. Некоторые авторы [69] сообщают о высоком риске местного рецидива (приблизительно 50%) в среднем в течение 53 мес (диапазон 10—115 мес) и росте опухоли со скоростью 34 мм/год (диапазон 1—10,8 мм). «Золотым» стандартом лечения небольших опухолей является открытая и лапароскопическая частичная нефрэктомия. В настоящее время используются также альтернативные методы — криотерапия и радиочастотная аблация [70]. Последние методы могут повлиять на результат патоморфологического диагноза, хотя, по некоторым данным, патоморфологический диагноз после первого цикла криотерапии приблизительно в 91% случаев подтверждает результаты предварительной биопсии [71].

Феохромоцитома выявляется примерно у 26% пациентов с синдромом VHL [37]. У пациентов с очевидно спорадической феохромоцитомой в 3—11% случаев впоследствии выявляют мутацию VHL [10, 12, 13]. Феохромоцитома может быть первым проявлением синдрома [30, 72]. В большинстве случаев надпочечниковые феохромоцитомы при VHL-синдроме двусторонние (синхронные или метахронные) [37, 73]. Вненадпочечниковые феохромоцитомы встречаются примерно в 30% случаев [37, 74—76]. Феохромоцитомы как часть синдрома VHL имеют исключительно норадреналиновый фенотип. Биохимические маркеры опухоли могут помочь отличить VHL-ассоциированные феохромоцитомы от феохромоцитом при синдроме МЭН 2-го типа [75]. Различия в биохимическом фенотипе при VHL-синдроме и МЭН 2-го типа связаны с различной экспрессией тирозингидроксилазы (TH) — лимитирующего фермента синтеза катехоламинов, и фенилэтаноламин-N-метилтрансферазы (PNMT). У пациентов с синдромом VHL отмечена низкая экспрессия PNMT, преобразующей норадреналин в адреналин. Различия биохимического фенотипа также связаны с различиями хранения, транспорта и секреции катехоламинов [77]. МЭН 2-ассоциированные феохромоцитомы содержат более высокие концентрации катехоламинов из-за более выраженной экспрессии TH. VHL-ассоциированные феохромоцитомы, секретируют катехоламины непрерывно, тогда как при синдроме МЭН 2 отмечен эпизодический характер секреции. Это определяет и различия клинических проявлений двух синдромов. Например, пациенты с МЭН 2 чаще жалуются на кризовые подъемы АД [78]. Помимо генетических различий [26], регистрируется разная экспрессия эритропоэтина и его рецептора [79]. Кроме того, около 80% феохромоцитом бессимптомны и выявляются случайно при визуализирующих исследованиях. Низкая чувствительность некоторых радионуклидных методов визуализации может объясняться относительной нехваткой гранул хранения или уменьшенной экспрессией мембранного норадреналина или везикулярных моноаминных транспортеров [80]. Поэтому сцинтиграфия с 123 I-MIBG (метайодбензилгуанидином) часто не обнаруживает VHL-связанные надпочечниковые феохромоцитомы [81, 82]. ПЭТ с 6-18F-фтордопамином более чувствительный метод [36, 83]. Злокачественные феохромоцитомы с метастазами в легких, печени, костях, лимфоузлах редко встречаются при синдроме VHL [37, 74, 84—87]. Метастазы выявляются менее чем в 7% случаев [37]. К сожалению, в настоящее время нет четких признаков, позволяющих надежно отличить доброкачественную от злокачественной феохромоцитомы, хотя уже известно, что герминальная мутация гена SDHB, является в этом отношении точным маркером [86—88]. Выявление феохромоцитомы у пациентов с синдромом VHL особенно важно, учитывая высокую вероятность хирургических вмешательств по поводу других опухолей (гемангиобластом ЦНС и др.). Невыявленная феохромоцитома при других вмешательствах может привести к опасным для жизни гипертоническим кризам. Более 70% феохромоцитом у детей являются VHL-ассоциированными. Каждому пациенту с VHL-синдромом и подтвержденной феохромоцитомой до оперативного лечения необходимо проводить ПЭТ с 6-18F-фтордопамином или сцинтиграфию с 123 I-MIBG для выявления вненадпочечниковой феохромоцитомы или метастазов [89]. Лечение феохромоцитомы оперативное. В то же время 6-месячная терапия ингибиторами тирозинкиназы приводит к уменьшению опухоли на 21% и сокращению уровня норметанефринов и хромогранина А в плазме [90] (рис. 4). Рисунок 4. Двусторонняя феохромоцитома и поликистоз почек.
Цистаденомы эпидидимуса — доброкачественные опухоли, которые могут быть двусторонними [48]. Чаще они имеют около 2 см в диаметре, могут распространяться на семенной канатик, приводя к бесплодию. Хирургическое лечение обычно не проводят; необходимо наблюдение [92].
У 35—75% пациентов с синдромом VHL имеются доброкачественные кисты и микрокистозные аденомы поджелудочной железы [93—96]. По данным КТ, у 17% пациентов выявляются панкреатические нейроэндокринные опухоли [95, 97, 98]. Из 633 пациентов с VHL-синдромом у 108 (17%) обнаруживались панкреатические эндокринные опухоли, и у 9 (8%) были выявлены метастазы [99]. Метастазирование более вероятно, если размеры опухоли превышают 3 см. У 78% пациентов с метастазами (7 из 9) мутация локализовалась в экзоне 3 гена VHL, и удвоение массы опухоли в среднем происходило за 337 дней. Таким пациентам необходимо оперативное лечение. Если размеры опухоли меньше 3 см, она растет медленно, а мутация локализована не в экзоне 3, то можно ограничиться наблюдением за пациентом. Средний возраст диагностики нейроэндокринных опухолей поджелудочной железы — 35 лет. Панкреатические кисты встречаются у больных начиная с 15 лет и чаще являются бессимптомными. В зависимости от размера и местоположения клинические симптомы могут быть вызваны обструкцией желчных путей и/или ферментной недостаточностью. В таких случаях устанавливают желчный стент и/или назначают ферментные препараты.
Гормонально-активные нейроэндокринные опухоли встречаются редко [95, 100]. По данным последних исследований, смертность при панкреатических эндокринных опухолях составляет 6%. В 60% случаев при сцинтиграфии обнаруживают рецепторы соматостатина; злокачественные опухоли выявлялись в 58% случаев [101].
Опухоли внутреннего уха располагаются в лабиринте, под твердой мозговой оболочкой на задней поверхности пирамиды височной кости [102—104]. Эти опухоли практически не метастазируют [105]. Симптомы включают потерю слуха, звон в ушах, головокружение и/или парез лицевого нерва [106, 107]. Таким образом, пациентам с мутацией VHL абсолютно показаны аудиологический осмотр, а также КТ или МРТ внутреннего уха с высокой разрешающей способностью, так как раннее хирургическое вмешательство способно сохранить слух. У пациентов с двусторонними опухолями, приводящими к глухоте, слух может быть восстановлен кохлеарным имплантом [108].
У 90% носителей мутации VHL к 60-летнему возрасту имеются те или иные клинические проявления синдрома [45]. На долгосрочный прогноз и смертность обычно влияет наличие гемангиобластом сетчатки и ЦНС, а также карциномы почки на поздних стадиях [31—33]. Таким образом, своевременное обследование и выявление патологии, ассоциированной с VHL-синдромом, является залогом успешного лечения и увеличения продолжительности жизни пациента (табл. 4).
Читайте также:
- Влияние экзогенных антиокислителей. Роль гормональных факторов в интоксикации кислородом
- Агрегация тромбоцитов при тяжелой декомпрессии. Активность лейкоцитов при декомпрессии
- Анатомия: Мышцы свода черепа. Фасции головы
- Истерический вариант развития слепого. Депривации у слепорожденных
- Синдром Жерлье (Gerlier)