Смерть-гибель ооцита. Причины апоптоза
Обновлено: 02.05.2024
Само название этого типа клеточной смерти – апоптоз, что в переводе с греческого означает «падающие листья», говорит о том, что он является такой же естественной и неотъемлемой чертой многоклеточного организма, как сезонная смена листвы для деревьев. Апоптоз запускается, когда клетка имеет серьезные повреждения, ведущие к нарушению ее функций: в результате слаженной работы специальных систем, необратимо повреждающих основные клеточные структуры, такая клетка заканчивает жизнь «самоубийством».
Все клетки многоклеточных существ несут в себе потенциальную способность к апоптозу, так же как японские самураи всю жизнь носят с собой меч. И если по каким-то причинам тонкий механизм апоптоза разлаживается, последствия для организма могут оказаться самыми катастрофическими. Например, раковые клетки, блокируя систему апоптоза, приобретают бессмертие. Поэтому изучение механизмов клеточной самоликвидации является важнейшим направлением современных биомедицинских исследований: раскрытие тайн апоптоза поможет в разработке новых лекарств для борьбы с самыми тяжелыми и трудноизлечимыми болезнями современности
Каждый день и каждый час в нашем организме погибают миллионы клеток. Отшелушиваются ороговевшие клетки покровного эпителия, быстро изнашиваются и гибнут клетки слизистой ткани, выстилающей пищеварительный тракт, лейкоциты – белые клетки крови, находят свою смерть в борьбе с патогенами… Но как наше тело избавляется от специализированных клеток, когда в результате накопившихся внутренних повреждений они становятся неспособными выполнять свои функции? Одним из самых парадоксальных и удивительных механизмов, контролирующих жизнеспособность многоклеточного организма, является апоптоз – клеточная самоликвидация.
Регулярная, генетически запрограммированная гибель отдельных клеток необходима для нормального функционирования организма в целом. Все клетки многоклеточных существ обладают аппаратом апоптоза, так же как японские самураи всю жизнь носят с собой меч. Однако у этого естественного процесса есть и обратная сторона: если по каким-то причинам тонкий механизм апоптоза разлаживается, последствия для организма могут оказаться самыми катастрофическими.
Нарушения в запуске апоптоза ведут к возникновению ряда серьезных заболеваний, в том числе аутоиммунных и онкологических. Например, раковые клетки, блокируя систему апоптоза, приобретают бессмертие. Поэтому изучение механизмов клеточной самоликвидации является важнейшим направлением современных биомедицинских исследований: раскрытие тайн апоптоза поможет в разработке новых лекарств для борьбы с самыми тяжелыми и трудноизлечимыми болезнями современности.
Ферменты-киллеры
Итак, клетка выполнила свои функции, «постарела» и готова к самоуничтожению во благо всему организму. Кто же выполняет это «заказное» самоубийство?
Оказывается, в этом «детективе» про апоптоз имеются и свои затаившиеся киллеры. В этой роли выступают особые ферменты – каспазы, имеющиеся в каждой клетке (Salvesen, 2002; Nicholson, 1999; Lavrik et al., 2005). Обычно каспазы присутствуют в клеточной цитоплазме в виде неактивных предшественников (прокаспаз). Прокаспазы не проявляют никакой активности, мирно сосуществуя в клетке вместе с другими белками, однако при поступлении сигнала на самоуничтожение они превращаются в настоящие белки-убийцы.
«Смена имиджа» безобидных прокаспаз происходит так: белок расщепляется на три фрагмента, один из которых (продомен) отщепляется, а остальные соединяются с двумя аналогичными фрагментами другой прокаспазы. Благодаря такой структурной перестройке образуется активный гетеротетрамер каспазы, в котором аминокислоты формируют центр фермента, выполняющий каталитическую функцию (Salvesen, 2002).
Образовавшиеся активные каспазы наконец показывают свое настоящее лицо: они начинают расщеплять все белки, которые содержат остатки аминокислоты аспарагина (при условии, что рядом располагаются определенным образом остатки еще трех других аминокислот). В результате такой «подрывной» деятельности в клетке оказываются поврежденными сотни белков. К числу наиболее известных мишеней каспаз относятся белки цитоскелета (структурного каркаса клетки); белки, отвечающие за репарацию (восстановление) поврежденной ДНК; структурные белки оболочки клеточного ядра, а также ряд других жизненно важных белков. Все это приводит к нарушению всех процессов жизнедеятельности клетки.
В то же время каспазы активируют ряд белков, которые участвуют в выполнении программы самоликвидации. Например, белка, который разрезает ДНК на большие фрагменты, – этот процесс, после которого целостность ДНК необратимо уничтожается, является характерной чертой апоптоза.
Сигнал на запуск
Но каким же образом клетка узнает, что ей пора самоликвидироваться? Кто и как дает указания киллерам-каспазам?

Имеется два основных пути, по которым передаются апоптопические сигналы в виде клеточных регуляторов, таких как гормоны, антигены, моноклональные антитела и другие молекулы. Это митохондриальный (или внутренний) путь, а также через особые трансмембранные белки – так называемые рецепторы смерти (DR, от англ. death receptor). В обоих случаях для запуска апоптоза должны образоваться особые инициаторные апоптотические комплексы. Затем происходит активация так называемых инициаторных каспаз, которые, в свою очередь, активируют эффекторные (разрушающие клеточные структуры) каспазы, о которых упоминалось выше (Nicholson, 1999).
Митохондриальный путь инициируется в результате интенсивного воздействия на клетку ряда повреждающих факторов. Однако каким образом эти повреждения трансформируются в митохондриальный апоптотический сигнал, пока в деталях не установлено. Тем не менее достоверно известно, что первым шагом на этом пути является выход из митохондрий («энергетических фабрик» клетки) цитохрома С – небольшого белка, содержащего комплекс с железом, который является компонентом митохондриальной дыхательной цепи (Green et al., 2004).
Выход цитохрома С инициирует образование в цитоплазме клетки крупного белкового комплекса – апоптосомы, в которую, помимо самого митохондриального белка, входят прокаспаза-9 и белок АПАФ-1. Именно апоптосома и является настоящим «мафиозным боссом» митохондриального пути апоптоза, который дает сигнал киллерам-каспазам.
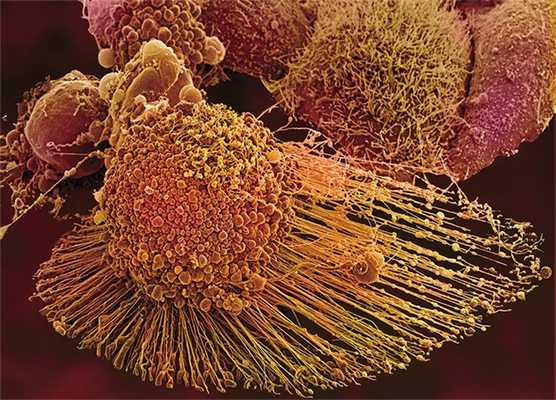
Речь идет об очень интересном явлении – самоактивации прокаспазы. Такое может произойти лишь в том случае, когда две молекулы этого белка, ориентированные определенным образом относительно друг друга, образуют димер. Именно такие уникальные пространственные условия, необходимые для димеризации и каталитической активации фермента, и предоставляет прокаспазе-9 апоптосома. Образовавшаяся в результате активная каспаза-9 расщепляет эффекторные каспазы (каспазу-3 и каспазу-7), а дальше все происходит по стандартной схеме апоптоза (Green et al., 2004).
В случае рецептор-зависимого сигнального пути инициация апоптоза начинается с другого белкового комплекса, который образуется непосредственно на самом рецепторе смерти (Krammer et al., 2007; Lavrik et al., 2005).

К настоящему времени семейство таких рецепторов включает шесть представителей, в том числе рецептор такого широко известного белка, как фактор некроза опухоли. Все рецепторы смерти имеют одинаковый фрагмент из 80 аминокислот – так называемый домен смерти, расположенный на белковом «хвостике», выходящем в цитоплазму клетки. Такой же аминокислотный фрагмент имеет и белок-адаптер FADD, находящийся в цитоплазме клетки. Домены смерти могут взаимодействовать между собой с образованием устойчивой связи; FADD, в свою очередь, способен присоединять к себе прокаспазу.
Вся цепь событий по образованию апоптотического комплекса запускается лигандом смерти – белком-агонистом, способным специфично связываться с рецептором смерти. Синтез (и, соответственно, рост концентрации) таких молекул в клетке стимулируется каскадом процессов, возникающих в ответ на повреждение клетки. В результате, благодаря посредничеству FADD, на рецепторе образуется комплекс DISC (от англ. death-inducing signaling complex), что в дословном переводе означает «сигнальный комплекс, инициирующий гибель». Именно в этом комплексе, как и в апоптосоме, происходит самоактивация прокаспазы-8, которая, в свою очередь, активирует эффекторные каспазы (каспазу-3 и каспазу-7) и инициирует клеточную гибель (Lavrik et al., 2005; Krammer et al., 2007). Собственно говоря, на этом различия между запуском двух сигнальных путей апоптоза заканчиваются.
Жить или не жить?
Нужно отметить, что любая клетка организма постоянно подвергается многочисленным повреждающим воздействиям, таким как радиационное излучение разных типов, разнообразные химические агенты, недостаток питательных веществ и т. п. К счастью для нас, для полноценной инициации клеточной гибели необходимо сравнительно сильное воздействие. На страже апоптотических путей стоят специфические механизмы, играющие роль «регулировщиков движения». Эту роль играют особые белки XIAPs и FLIP (Lavrik et al., 2005).

Белки XIAPs ингибируют каспазу-9, которая активируется вследствие развертывания митохондриального пути. Связываясь с активным центром каспазы, они не дают «киллеру» выполнять свою работу. Однако с помощью этих белков клетке удается заблокировать лишь небольшое число активных каспаз. Если же концентрация активных каспаз превышает некий пороговый уровень, то белков XIAPs становится недостаточно, и процесс апоптоза остановить уже невозможно.
В случае рецепторзависимого сигнального пути апоптоза ингибитором активации прокаспазы-8 служит близкий ей по структуре белок FLIP. Молекулы этого белка также могут связываться с апоптическим комплексом DISC, конкурируя за «место» с молекулами прокаспазы, – при повышенной концентрации в цитоплазме они блокируют все возможные «места» такого связывания (Krammer et al., 2007). В результате прокаспаза-8 не может быть активирована, и апоптоз не запускается.
Нарушения в уровне экспрессии как про- так и антиапоптотических белков может привести к серьезным отклонениям от обычного образа жизни клетки. Так, повышенный уровень экспрессии белков XIAPs и FLIP имеют многие раковые клетки. Выбрав курс на собственное бессмертие, в конечном счете они приводят к гибели все многоклеточное «сообщество» организма.
Итак, в отличие от голливудского детектива, в истории про апоптоз нет главного действующего лица: своевременное уничтожение поврежденных клеток и в итоге – жизнеспособность организма зависит от слаженной цепочки событий, в которой участвует множество различных белковых молекул.
И здесь очень важны количественные показатели, такие как концентрация. Сегодня изучением того, как влияет на инициацию и дальнейший ход апоптоза уровень содержания в клетке различных молекул, занимается одна из передовых областей современной науки – системная биология (Bentele et al., 2004). Основной ее постулат заключается в том, что протекание сложных процессов внутри клетки можно понять, лишь учитывая максимально большое число клеточных параметров. Для этого на основе экспериментальных данных создается компьютерная модель, которая учитывает действие множества факторов. Полученные таким образом предсказания о ходе основных клеточных процессов могут использоваться в борьбе с препятствиями человечества на пути к долгой и здоровой жизни.
Lavrik I. N., Golks A., Krammer P. H. Caspases: Pharmacological manipulation of cell death // J. Clin. Invest. 2005. V. 115, N 10. P. 2665—2672.
Krammer P. H., Arnold R., Lavrik I. N. Life and death in peripheral T cells // Nat. Rev. Immunol. 2007. V. 7. P. 532—542.
Green D. R. and Kroemer G. The pathophysiology of mitochondrial cell death // Science. 2004. V. 305. P. 626—629.
Смерть-гибель ооцита. Причины апоптоза
Во время эмбриогенеза в развивающихся гонадах закладывается избыточное количество половых клеток и в течение всего онтогенеза наблюдается гибель ооцитов, достигающая 99,9% популяции. Апоптоз является одним из естественных путей редукции популяции клеток яичников. Современные данные о роли апоптоза в функционировании яичников не только в норме, но и при развитии опухолевого процесса играют огромную роль в достижении глобальной цели: предотвращение и возможности профилактики целого ряда заболеваний репродуктивной системы [2, 12].
Начиная с 2000 г., апоптозу в яичниках посвящено огромное количество работ 3. Было доказано на культуре клеток яичника, что апоптоз - основной механизм атрезии фолликулов [3, 6, 22], а также запрограммированная клеточная гибель происходит в эпителии органа в момент овуляции и в желтом теле [6, 11].
Клетками, более всего подверженными апоптозу, связанному с атрезией фолликулов, являются клетки гранулезы [3, 10, 11]. Позднее апоптоз обнаруживается в клетках теки (Tilly, 2004), хотя некоторые авторы считают, что апоптоз характерен, как правило, для клеток гранулезы, но не для клеток теки [24]. Есть доказательства, что атрезии фолликулов на всех стадиях развития предшествует апоптоз ооцитов [24, 26].
Возможно, начальные проявления апоп-тоза и гибель ооцита, а также выживаемость клеток гранулезы зависит от образования межклеточных контактов и экспрессии коннексина-43, который обеспечивает образование щелевых контактов и передачу сигналов, ведущих к апоптозу [18].
Японскими учеными изучались возможности апоптоза в доминантных фолликулах перед овуляцией в период 2-й и 3-й фолликулярных волн в связи с возрастом фолликулов. Исследования показали, что клетки, подвергнутые апоптозу, встречаются на 18 день доминантных фолликулов перед овуляцией в период, как второй, так и третьей фолликулярных волн. На программированную клеточную гибель в яичниках влияют не только гены регуляторы, но и различные цитокины и гормоны [30].
По данным английских исследователей апоптоз лютеинизированных клеток грану-лезы и клеток яйценосного бугорка зависит от возраста женщины. Доказано, что в возрасте 31-35 лет частота апоптоза этих клеток значительно выше, чем до 31 года. Сделан вывод о присутствующем в фолликулярной жидкости прогестероне, играющим защитную роль и уменьшающим частоту апоптоза лютеинизированных клеток гранулезы [11].
По данным Сергеева (2008г.) гестагены косвенно или напрямую взаимодействуют с системами регуляции пролиферации на всех известных уровнях [26]. Однако это действие в большой мере зависит от дозы, времени, кофакторов действия, а также фазы клеточного цикла и, возможно, от других, не определенных пока факторов. Показано, что гестагены могут влиять на уровень активности некоторых факторов апоптоза, запуская каскады реакций. Прогестерон подавляет апоптоз гранулезных клеток в ранних фолликулах. Апоптоз гранулезных клеток связан с дисбалансом между прогестероном и эстрадиолом в фолликулярной жидкости [1, 2, 3, 26]. Уровень прогестерона и эстрадиола в фолликулярной жидкости меняется независимо от концентрации в сыворотке в течение менструального цикла, но зависит от размера фолликула и степени атрезии [3]. Концентрация эстрадиола и прогестерона и уровень апоп-тоза в доминантных фолликулах меняются прямо противоположно в сравнении со средними фолликулами, подтверждая мнение о том, что доминантные фолликулы подавляют рост субдоминантных [2, 3].
Прогестины также влияют на активность некоторых ферментов клеток-мишеней, задействованных в процессах проведения сигнала гибели клеток, модулируя ответ клетки на апоптический сигнал. Каспаза-3 - фермент, индуцируемый прогестероном, является компонентом проведения сигнала рецепторной системы Fas/FasL, которой отводится роль в реализации апоптоза, в частности в клетках ткани яичника. Прогестерон ингибирует апоптоз в клетках желтого тела путем подавления Fas и активности каспазы-3. Значение этой системы показано в реализации гибели различных опухолевых клеток, в том числе клеток опухолей репродуктивной системы, а также нормальных клеток яичников [8, 27, 28, 29]. Рост и атрезия малых фолликулов у новорожденных и половозрелых мышей, по данным некоторых авторов, не связана с активностью каспазы-3 [27, 29]. Согласно другому мнению в яичниках позвоночных апоптоз опосредуется именно каспазами, благодаря чему обеспечивается удаление избыточных или нежизнеспособных зародышевых и гранулезных клеток, при этом чрезмерность апоптоза и атрезия фолликулов могут отрицательно влиять на фертильность [16]. Нарушения апоптоза вовлечены также в патогенез хронической ановуляции и овари-альной дисфункции [3, 24]. Таким образом доказано, что половые стероиды, воздействуя на экспрессию про- и антиапоптозных белков, могут контролировать процессы апоптоза в репродуктивной системе.
К. Kugu и ряд других авторов идентифицировали в яичниках белки bcl, bax и р53 [20, 21, 23, 27]. Семейству белков bcl-2 принадлежит важная роль в регуляции апоп-тоза. Данные факторы принимают участие в формировании ионных каналов, а также проявляют специфическую ферментативную активность или являются транскрипционными факторами [3, 14]. Среди них bcl-2 и bcl-xL предотвращают апоптоз, в то время как bax и bcl-xS индуцируют клеточную смерть. Иммуногистохимически доказано, что bcl-2 экспрессируется в середине люте-иновой фазы в лютеиновых клетках, но не в регрессирующем желтом теле. Наиболее высокие уровни bax, наоборот, определяются в регрессирующем желтом теле [14]. Прогестерон в концентрации 10-8 М повышает экспрессию фактора bcl-2, снижает экспрессию фактора bax и снижает активность участвующих в реализации апоптозного сигнала протеаз, в частности, каспазы-3 [3].
Важным регулятором развития фолликула и желтого тела является р53 [1]. Апоптоз клеток гранулезы связан с экспрессией р53, который запускает процесс апоптоза только в гранулезных клетках зрелого преовуля-торного фолликула [23]. Однако согласно другим исследованиям, экспрессия р53 увеличивается одновременно с ростом количества апоптотических клеток гранулезы и тека-клеток, а также изменяется при созревании фолликула и находится в обратной зависимости от уровня хорионического го-надотропина, так как ХГ блокирует апоптоз и подавляет индукцию р53 [1]. Сопутствующая аккумуляции органелл стероидогенеза в перинуклеарном пространстве, способность протеосом к дегенерации совпадает с аккумуляцией р53 в ядре [3, 23].
У самок млекопитающих число ооцитов в яичниках, как известно, уменьшается посредством апоптоза на протяжении всей жизни. Авторы данной гипотезы, согласно которой запас питательных веществ у особи регулирует жизнеспособность ооцитов, показали, что пентозно-фосфатный путь генерации NADPH играет решающую роль в выживании генеративных клеток, а мишенью этой регуляции служит каспаза-2, которая вызывает гибель ооцитов. Опосредованное пентозо-фосфатным путем ингибирование гибели ооцитов обусловлено подавлением фосфорилирования каспазы-2 посредством Ca-кальмодулин-зависимой протеинкиназы II. Сделан вывод о том, что истощение питательных веществ для ооцита приводит к неспособности генерирования NADPH и апоптозу половых клеток. Следовательно, существует прямая связь между метаболизмом ооцита, Ca-кальмодулин-зависимой протеинкиназой II и каспазой-2 [16, 27, 29].
Опыты in vitro на яичниках крыс позволили сделать вывод о том, что перекись водорода зависимо от концентрации и времени вызывала сморщивание незрелых и зрелых ооцитов крысы, образование пузырьков в плазматической мембране и дегенеративные изменения, характерные для апопто-за. Также, в 3 раза повышалась экспрессия белка Bax, в 2,5 раза увеличивалась активность каспазы-3 и повышалась фрагментация ДНК [16, 20]. Таким образом, перекись водорода стимулирует апоптоз в половых клетках.
Наиболее значимые паракринные регуляторы пролиферативных процессов в клетках эпидермального происхождения представлены семействами инсулиноподобного фактора роста (IGF), эпидермального фактора роста (EGF) и трансформирующего фактора роста в (TGF-P) [15]. Семейство IGF стимулирует пролиферацию и дифференциров-ку, являясь антиапоптическими факторами [17]. Показано, что повышение экспрессии рецепторов группы EGF в клетках нормального эпителия яичников положительно коррелирует с введением препаратов гестагенов [15, 24]. TGF-в рассматривается в качестве мультифункционального фактора роста, является ингибитором роста для клеток различных типов, в то время как TGF-a играет важную роль в качестве стимулирующего фактора роста [5].
По данным болгарских исследователей ингибин А снижает интенсивность апоп-тоза и стимулирует образование эстрадио-ла гранулезными клетками [10]. Аполи-попротеин Е является предположительно аутокринным регулятором компартмента текальных клеток яичников крыс, стимулируя апоптоз текальных и интерстициальных клеток яичников крыс, подтверждаемый увеличением числа пикнотических клеток, TUNEL-положительных клеток и лестничный электорофореграммами ДНК [3]. Базовым моментом фрагментации ДНК в поверхностных эпителиальных клетках яичника в норме при овуляции является воздействие прогестерона [28]. Фрагментация
ДНК, апоптоз происходят в эпителиальных клетках в месте овуляции. Клетки, содержащие фрагментированную ДНК, экспрессируют р53 [21, 23].
В условиях ex vivo с применением непрямой иммунофлуоресценции изучалась экспрессия тканевого ингибитора - 4 ме-таллопротеиназ (ТИМП-4) в яичниках мышей. У интактных неполовозрелых мышей ТИМП-4 был локализован в теке, антраль-ных фолликулах, в предовуляторных фолликулах и в прилегающей строме яичников. При воздействии ХГЧ отмечалась локализация ТИМП-4 в гранулезе при лютеинизации и дальнейшая устойчивая экспрессия в желтом теле. У половозрелых мышей ТИМП-4 был локализован в желтых телах, сохранившихся с предшествующих циклов, в теке предовуляторных фолликулов и, в меньшей степени, во вновь образуемых желтых телах. Экспрессия ТИМП-4 повышалась при лю-теинизации гранулезы, но существенно не изменялась на стадиях эволюции желтых тел. На основании этих данных сделан вывод о ведущей роли ТИМП-4 в поддержании функций желтого тела на всех этапах его эволюции [7].
Австралийскими морфологами впервые в 2005 году показано, что секретируемые ооцитом факторы, а также костные морфогенетические белки предотвращают апоптоз фолликулярных клеток в яичнике у млекопитающих [15].
В последнее время появилось много работ по изучению процесса апоптоза в опухолях [4, 25]. Лю Б.Н. подтверждает представление о том, что «стартовым моментом» для индукции канцерогенеза и апоптоза является дисфункция клеточного дыхания, приводящая к окислительному стрессу сначала в митохондриях, затем в цитоплазме и клетке в целом. Избыточное образование активных форм кислорода, перекисей липи-дов и белков, рассматриваемые как сигнальные молекулы, создает дисбаланс, величина которого возрастает в стареющих клетках, но еще более - в опухолевых и апоптиче-ских. Далее эти сигнальные молекулы играют ключевую роль на всех этапах работы исполнительных звеньев (теломераз, онко-белков, факторов транскрипции, протеин-киназ, эндонуклеаз, каспаз и др.). Сурвивин принадлежит к семейству белков - ингибиторов апоптоза. Показана корреляция между экспрессией сурвивина и выраженностью апоптоза в клеточных линиях рака яичников у человека [25]. Другие работы посвящены роли противоопухолевых препаратов в индукции апоптоза в яичниках [4]. Так ци-сплатин индуцирует апоптоз, предотвратить который возможно с помощью киназ ERK1/2. Ксанторризол оказывает антипролиферативное действие на клетки MCF-7 индуцируя апоптоз путем модуляции уровней белков bcl-2, p53 и PARP-1 [14, 20, 21, 23]. Альтерация регулирования этого процесса может привести к снижению клеточной смерти и развитию неопроцесса. Содержание bcl-2 выше в нормальных тканях, в то же время высокий уровень bax и bcl-xS отмечается в карциноме [9, 14, 25, 28]. Доказано, что в развитии рака яичника играет роль семейство генов-регуляторов апоптоза bcl-2, но не р53 [25]. Накопление же белка р53 в клетках приводит не к гибели раковых клеток, а к анти-р53-антителопродукции и к появлению соответствующих антител в сыворотке крови [21].
Японские исследователи изучали влияние теплового шока на функции клеток грануле-зы крыс. Тепловой шок приводил к уменьшению количества рецепторов ФСГ и ЛГ клеток гранулезы антральных фолликулов и снижал эстрогенную активность созревающих фолликулов. Клетки гранулезы в условиях теплового шока проявляют повышенную чувствительность к апоптозу. Система генов bcl-2/bax не связана с апоп-тозом клеток гранулезы, стимулированным тепловым шоком [26]. Изучалось влияние иммобилизационного стресса, антиоксидантов, а также комплексного воздействия этих факторов на апоптоз [9]. Установлено влияние ключевых гормонов стресса - глюкокортикоидов на экспрессию генов апоптоза в неонатальном периоде. В процессе возрастной инволюции усиливается перекисное окисление липидов, которое приводит к повышению уровня апоптоза. Этот процесс, вероятно, отличается от физиологического апоптоза по биохимическим характеристикам. С другой стороны, снижение уровня апоптоза может привести к канцерогенезу. Витамин Е, обладая выраженным антиоксидантным действием, принимает участие в регуляции апоптоза, в том числе в гонадах [14].
По данным отечественых ученых на клеточную пролиферацию и апоптоз могут влиять и аминокислоты. Так, гистидин, аспарагиновая кислота, валин, треонин, пролин, метионин и др. эффективно снижают частоту апоптоза в тканях различного генеза. Полученные данные указывают на необходимость использования пептидных биорегуляторов для коррекции возрастной патологии, связанной с дисбалансом обновления тканей [15, 24]. По данным Lea R.G. (2006) ограничение материнского питания во время раннего периода беременности влияет на регуляторы апоптоза в яичниках, повышая экспрессию некоторых факторов [19].
Таким образом, апоптоз представляет собой процесс, неотъемлемый от функционирования яичника. Он отвечает за развитие доминантного фолликула и желтого тела, фолликулярную атрезию и овуляцию. Нарушения в процессе апоптоза ведут к развитию многих патологических состояний, включающих ановуляцию, синдром поли-кистозных яичников, преждевременную недостаточность яичников, а также неопроцессы в гонадах [1]. Задачей будущих исследований является изучение возможности медикаментозного влияния на апоптоз с целью коррекции патологических изменений в женской половой железе, а также возможности повышения положительных исходов программ ЭКО.
1. Антонеева И.И., Петров С.Б. // Онкология. - 2008. - Т.10. №2. С. 234.
2. Боярский К.Ю. // Пробл. репродукции. - 2006.- №4. С. 26.
3. Дубровина С.О. // Рос. вестн. акушера-гинеколога.- 2006.- N 3. C. 33.
4. Манухин И.Б., Высоцкий М.М., Горюн С.В. и др. // Пробл. репродукции.- 2006. С. 71.
5. Bauer Georg. // Int. J. Radiat. Biol.-2007.- N 11-12. P. 873.
6. Brodowska A., Laszczynska M., Starczewski A. // Post. biol. komorki.- 2006. - N 1. P. 35.
7. Bu S., Cao Chenfu, Yang Yongjun. //Reproduction.- 2006.- N 6. P. 1099.
8. Campagnoli C., Abba С., Ambroggio S. et al. // J. Steroid Biochem. Mol. Biol. - 2007. - Vol.97. P. 441.
9. Cheah Yew Hoong, Azimahtol Hawariah Lope Pihie, Abdullah Noor Rain. // Anticancer Res.- 2006.- N 6B. Р. 4527.
10. Denkova R., Bourneva V., Zvetkova E. et al. // Acta morphol. et anthropol.- 2005.-N 10. P. 95.
11. Dineva J., Nikolov G., Vangelov I. et al. // Докл. Бълг. АН.- 2004. - N 8. P. 113.
12. Fadeel B., Orrenius S. // J. Int. Med. - 2005.- Vol. 258, №6. - P. 479-517.
13. Gastal E.L., Gastal M.O., Ginther O.J. //Reproduction. - 2006. - №4. P. 699.
14. Havelka P., Oborna I., Brezinova J. etal. // Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub. - 2005. - Vol. 149, №2. P. 303.
15. Hussein, Tamer G., Froiland David A., Amato Fred. et al. // J. Cell Sci. - 2005.- N 22. P. 5257.
16. Johnson A., Bridgham Jamie T. //Reproduction. - 2002. - V. 124, N 1. P.19.
17. Kooijman R. // Cytokine Growth Factor Rev. - 2006. - Vol.17, №4. P. 305.
18. Krysko Dmitri V., Mussche Sylvie, Leybaert Luc, D´Herde Katharina. // J. Histochem. and Cytochem. - 2004. - N 9. P. 1199.
19. Lea R.G., Andrade L.P., Rae M.T. et al. // Reproduction.- 2006.- N 1. P. 113.
20. Liszewska E., Rekawiecki R., Kotwica J. // Prostaglandins Other Lipid Mediat. - 2005. - Vol. 78, № 1. P.67.
21. Lumachi F. // Anticancer Res. - 2006. - Vol. 26, №2A. P. 1305.
22. Peter A.T., Dhanasekaran N. // Reprod.Domest. Anim.- 2003. - N 3 P. 209.
23. Rivera A., Mavila A., Bayless K.J. et al. // Cell. and Mol. Life Sci.- 2006. - N 12. P. 1425.
24. Sato Eimei, Kimura Naoko, Yokoo Masaki et al. // Microsc. Res. and Techn. - 2006. - V.69, N 6. P. 427.
25. Sato Takaji, Aoki Noriko, Komaki Rei et al. // Yakugaku zasshi.- 2005.- P. 100.
26. Shimizu Takashi, Ohshima Izumi, Ozawa Manabu et al. // Reproduction.- 2005.- № 4. P. 463.
27. Slot K.A., Voorendt M., Boer-Brouwer M. et al. // J. Endocrinol. - 2006. - Vol. 188. №2. P. 179.
28. Terry K.L., De Vivo I., Titus-Ernstoff L. // Am. J. Epidemiol. - 2005. - Vol. 161. №5. P. 442.
29. Xu Q., Takekida S., Ohara N. et. а! // J. Clin. Endocrinol. Metab. - 2005. - Vol. 90. №2. P. 953.
30. Yasuda K., Hagiwara E., Takeuchi A. et al. // Zool. Sci. - 2005. - №2. P. 237.
Ооциты являются, возможно, важнейшим лимитирующим фактором репродукции человека, потому что в течение репродуктивного периода жизни женщины всего около 300-400 яйцеклеток овулируют, т.е. свыше 99,9% ооцитов подвергается запрограммированной клеточной гибели, или апоптозу, без возможности созревания, овуляции и оплодотворения.
На мышах показано, что первая волна апоптоза происходит на этапе герминативного синцития, содержащего оогонии. Предполагается, что сходный процесс происходит у человеческого эмбриона женского пола. Количество ооцитов у человека достигает максимального значения (7 млн) в середине гестации. Впоследствии те ооциты, которые не приобрели оболочку из гранулезных клеток, подвергаются апоптозу. Далее растущие фолликулы подвергаются атрезии по мере достижения ими антральной стадии, так что к моменту рождения у девочки присутствует 300-400 тыс. фолликулов, а к пубертату — всего около 200 тыс. .
Хотя апоптоз — наиболее распространенный исход для клеток, инициирующие факторы и механизмы апоптоза могут различаться в зависимости от того, на какой стадии развития клетки он происходит. В частности, апоптоз первичных ооцитов, происходящий до образования примордиальных фолликулов, является автономным, т.е. реализующимся за счет внутренних механизмов самого ооцита, в то время как ооциты растущих преантральных и антральных фолликулов гибнут вторично, в результате атрезии фолликулов, вызванной апоптозом гранулезных клеток.
Для объяснения столь высокой степени коэффициента гибели ооцитов было предложено множество гипотез, в том числе основанных на предположении, что элиминация герминативных клеток, содержащих хромосомные и иные дефекты, является приоритетной задачей видов. Тем не менее ни одна из гипотез не доказана, поэтому вопрос до сих пор остается открытым.
Влияние окружающей среды на ооциты. Кроме апоптоза, являющегося частью естественного развития, истощение пула ооцитов может стимулироваться факторами окружающей среды и генетической предрасположенностью, что клинически зачастую проявляется синдромами ПЯН или ранней менопаузы. К ооцитотоксичным факторам окружающей среды относятся хлорсодержащие соединения в составе пестицидов и компонентов промышленных производств, полициклические ароматические углеводороды табачного дыма и выхлопных газов, химиотерапевтические препараты (например, доксорубицин), которые инициируют церамидный путь апоптоза (церамид — липид, образующийся под действием кислой сфингомиелиназы).
Повреждающий эффект церамида подтверждается защитными свойствами его метаболита и антагониста, сфингозин-1-фосфата, который предотвращает апоптоз ооцитов при химиотерапии.

Генетические причины апоптоза ооцитов
Генетические модели мышей апоптоза ооцитов демонстрируют различные фенотипы: от субфертильности (т.е. небольшое количество потомства в помете или нечастые пометы) до ПЯН и бесплодия вследствие врожденной атрезии яичников. Генетические дефекты могут в основном быть обусловлены структурными аномалиями хромосом или анеуплоидией, дефектами Х-инактивации; мутациями отдельных генов, необходимых для оогенеза, фолликулогенеза или гонадогенеза; генными дефектами, затрагивающими механизмы апоптоза в яичнике. Примеры последних приведены в этой главе, остальные описаны в других главах.
Следует подчеркнуть, что апоптоз — общий финал при развитии дефектов, нарушающих оогенез.
Как и в случае с соматическими клетками, для поддержания «нормальной» скорости истощения ооцитов в них должны адекватно экспрессироваться про- и антиапоптотические белки. К защитным факторам относятся экспрессируемые в ооцитах и гранулезных клетках белки Вс1-2 и Bcl-х. Хотя у Bcl-2-дефицитных мышей отмечают нормальное количество потомства в помете, у них снижается количество примордиальных фолликулов и повышается число дефектных примордиальных фолликулов, содержащих гибнущие ооциты или не содержащих их вовсе.
У гипоморфных по Bcl-х мышей также существенно снижено количество примордиальных и первичных фолликулов, но патофенотип у них более выражен и фертильность существенно снижена. Таким образом, эти два гена необходимы для поддержания пула ооцитов и архитектоники фолликулов. В противоположность этому направленная гиперэкспрессия Вс1-2 гранулезными клетками проявляется интенсификацией фолликулогенеза, увеличением количества потомства в помете и возрастным повышением частоты развития опухолей из гранулезных клеток.
Самки мышей с дефицитом Вах фертильные и имеют больше примордиальных фолликулов по сравнению с мышами дикого типа, даже в позднем возрасте. Более того, они способны к суперовуляции в ответ на введение экзогенных гонадотропинов, причем такие ооциты дают жизнеспособные эмбрионы даже в позднем репродуктивном возрасте. Такие же результаты были получены на моделях мышей с делецией проапоптотического гена капсазы-2. Кроме того, в отсутствие капсазы-2 ооциты при воздействии на них химиотерапевтического препарата доксорубицина апоптозу не подвергались.
Эти данные указывают на необходимость дальнейших исследований, способных приблизить нас к лучшему пониманию и потенциальному увеличению репродуктивного периода жизни женщины. И, что самое важное, понимание механизмов регуляции апоптоза в клетках млекопитающих необходимо для разработки химиотерапевтических и других фармакологических препаратов, не нарушающих функции ооцитов и фолликулов.
Преждевременная недостаточность яичников. Причины раннего бесплодия
Многие ключевые проблемы современной клинической репродуктивной медицины связаны с доступностью и функциональной полноценностью человеческих ооцитов и окружающих их фолликулов. Пока мы весьма далеки от понимания того, какая доля «идиопатического бесплодия» связана с ооцит-специфичными факторами, и еще дальше — от разработки методов терапии, ооцит-специфической терапии. Прогресс в лечении бесплодия, вызванного фактором ооцита, возможен только при условии лучшего понимания механизмов влияния возраста матери, эффектов окружающей среды и индивидуальной генетической предрасположенности на функции ооцита.
На моделях человека и мышей апоптоза ооцитов были получены различные фенотипы, варьирующие от субфертильности и ПЯН до врожденной атрезии яичников. Хотя эти клинические состояния рассматривают как отдельные нозологические формы с разными подходами к лечению, их генетическая основа может быть единой. В целом генетические дефекты могут быть обусловлены структурными или анеуплоидными хромосомными аномалиями, в том числе Х-хромосомы (например, синдром Шерешевского-Тернера), дефектами инактивации Х-хромосомы, мутациями или дефектами эпигенетической регуляции отдельных генов, необходимых для оогенеза, фолликулогенеза или гонадогенеза.
В частности, семейные или возникшие de novo генные мутации, затрагивающие любую стадию оогенеза, теоретически могут привести к целому спектру патологических состояний — от врожденной дисгенезии гонад до ПЯН или плохому качеству ооцитов, выявляемому при стимуляции яичников и ЭКО.
Из всех состояний, связанных с патологией ооцитов или фолликулов яичников, семейная форма поликистоза яичников (ПЯН) является самым излюбленным объектом для выявления генных мутаций человека. Семейные случаи ПЯН, обусловленные сбалансированной транслокацией, можно изучать с использованием цитогенетиче-ских и флюоресцентных методов гибридизации in situ (FISH).
К примеру, многие аутосомно-доминантные мутации FOXL2 — фактора транскрипции, экспрессируемого только в гранулезных клетках и клетках века глаза, становятся причиной синдрома ПЯН в сочетании с мальформацией век, называемого синдромом блефарофимоза-птоза-обратного эпикантуса.

Другая генетическая мутация, связанная с семейной гипергонадотропной недостаточностью гонад у людей, локализована на Х-сцепленном гене ВМР15. Более того, «критический регион» длинного плеча Х-хромосомы (Xql3-Xq26) предположительно содержит несколько генов, необходимых для оогенеза и развития яичников. Семейный вариант ПЯН также обусловлен мутацией гена, расположенного в этом «критическом регионе», а именно DIA, являющегося гомологом гена Drosophila diaphanous («прозрачный»), играющего важную роль в оогенезе плодовой мушки.
Учитывая значение мышиных гомологов генов FOXL2 и ВМР15, а также гена Drosophila diaphanous в оогенезе и фолликулогенезе, приведенные примеры иллюстрируют возможность интерпретации знаний, полученных в экспериментальных моделях, на клиническую практику.
Подобные генные мутации человека являются стихийными аналогами моделей направленного мутагенеза, демонстрирующих роль этих генов в репродукции человека in vivo. Более того, изучение биохимии белков, кодируемых этими генами, может приблизить нас к пониманию патогенеза и разработке перспективных методов терапии бедного ответа яичников на введение гонадотропинов, ПЯН и синдрома гиперстимуляции яичников.
В отдаленном (а может быть, и не столь уж отдаленном) будущем возможно (по мере идентификации генетических мутаций человека, приводящих к дисфункции яичников) создание программ генетического скрининга и консультирования, способных прогнозировать риск преждевременного старения ооцитов, что позволит молодым женщинам более эффективно планировать семью. Пациенток с бедным ответом яичников или риском развития синдрома гиперстимуляции яичников можно будет выявлять еще до проведения программ ВРТ.
Разработка достоверных предикторов этих состояний поможет не только оптимизировать исход лечебного цикла, но и консультировать супружеские пары относительно прогноза лечения, риска многоплодия и синдрома гиперстимуляции яичников.
Анеуплоидия. Нарушения расхождения хромосом в мейозе приводят к анеуплоидии, частота которой у человека увеличивается с возрастом женщины. Анеуплоидия, считающаяся важнейшей проблемой репродукции человека, встречается в 25-50% яйцеклеток человека, является причиной по меньшей мере 35% самопроизвольных абортов и около 4% мертворождений, являясь при этом ведущей генетической причиной врожденной умственной отсталости и дефектов физического развития.
Важнейшие факторы риска возникновения в ооцитах анеуплоидии у человека — возраст женщины и высокая модель рекомбинации в ходе мейоза, например, в тех участках, которые располагаются ближе к центромере или теломере. По сравнению с ооцитами мышей, у человека имеет место чрезвычайно высокая вариабельность в расположении узелков рекомбинации, что может объяснить высокую частоту анеуплоидии у человека. Однако, несмотря на то, что уязвимые локализации узелков рекомбинации считают основным фактором риска развития анеуплоидии у молодых женщин, они не объясняют возрастания частоты анеуплоидии с возрастом.
На самом деле связь между различными моделями рекомбинации и частотой анеуплоидии становится с возрастом все слабее. В связи с этим предполагают, что не зависящие от моделей рекомбинации факторы риска анеуплоидии для женщин старшего репродуктивного возраста пока еще неизвестны.
VIII Международная студенческая научная конференция Студенческий научный форум - 2016

Апоптоз - программированная клеточная гибель, энергетически зависимый, генетически контролируемый процесс, который запускается специфическими сигналами и избавляет организм от ослабленных, ненужных или повреждённых клеток. Ежедневно, примерно около 5% клеток организма подвергаются апоптозу, а их место занимают новые клетки. В процессе апоптоза клетка исчезает бесследно в течение 15-120 минут.
Запрограммированная клеточная гибель это биохимически специфический тип гибели клетки, который характеризуется активацией нелизосомных эндогенных эндонуклеаз, которые расщепляют ядерную ДНК на маленькие фрагменты. Морфологически апоптоз проявляется гибелью единичных, беспорядочно расположенных клеток, что сопровождается формированием округлых, окруженных мембраной телец (“апоптотические тельца”), которые тут же фагоцитируются окружающими клетками.
Апоптоз – энергозависимый процесс, посредством которого удаляются нежелательные и дефектные клетки организма. Он играет большую роль в морфогенезе и является механизмом постоянного контроля размеров органов. При снижении апоптоза происходит накопление клеток, пример – опухолевый рост. При увеличении апоптоза наблюдается прогрессивное уменьшение количества клеток в ткани, пример – атрофия.
Морфологические проявления апоптоза.
Апоптоз имеет свои отличительные морфологические признаки, как на светооптическом, так и на ультраструктурном уровне. При окраске гематоксилином и эозином апоптоз определяется в единичных клетках или небольших группах клеток. Апоптотические клетки выглядят как округлые или овальные скопления интенсивно эозинофильной цитоплазмы с плотными фрагментами ядерного хроматина. Поскольку сжатие клетки и формирование апоптотических телец происходит быстро и также быстро они фагоцитируются, распадаются или выбрасываются в просвет органа, то на гистологических препаратах он обнаруживается в случаях его значительной выраженности. К тому же апоптоз – в отличие от некроза – никогда не сопровождается воспалительной реакцией, что также затрудняет его гистологическое выявление.
Наиболее четко морфологические признаки выявляются при электронной микроскопии. Для клетки, подвергающейся апоптозу характерно:
Сжатие клетки. Клетка уменьшается в размерах; цитоплазма уплотняется; органеллы, которые выглядят относительно нормальными, располагаются более компактно. Предполагается, что нарушение формы и объема клетки происходит в результате активации в апоптотических клетках трансглютаминазы. Этот фермент вызывает прогрессивное образование перекрестных связей в цитоплазматических белках, что приводит к формированию своеобразной оболочки под клеточной мембраной, подобно ороговевающим клеткам эпителия.
Конденсация хроматина. Это наиболее характерное проявление апоптоза. Хроматин конденсируется по периферии, под мембраной ядра, при этом образуются четко очерченные плотные массы различной формы и размеров. Ядро же может разрываться на два или несколько фрагментов. Механизм конденсации хроматина изучен достаточно хорошо. Он обусловлен расщеплением ядерной ДНК в местах, связывающих отдельные нуклеосомы, что приводит к развитию большого количества фрагментов, в которых число пар оснований делится на 180-200. При электрофорезе фрагменты дают характерную картину лестницы. Эта картина отличается от таковой при некрозе клеток, где длина фрагментов ДНК варьирует.
Формирование в цитоплазме полостей и апоптотических телец. В апоптотической клетке первоначально формируются глубокие впячивания поверхности с образованием полостей, что приводит к фрагментации клетки и формированию окруженных мембраной апоптотических телец, состоящих из цитоплазмы и плотно расположенных органелл, с или без фрагментов ядра.
Фагоцитоз апоптотических телец. Фагоцитоз апоптотических клеток или телец осуществляется окружающими здоровыми клетками, или паренхиматозными, или макрофагами. Апоптотические тельца быстро разрушаются в лизосомах, а окружающие клетки либо мигрируют, либо делятся, чтобы заполнить освободившееся после гибели клетки пространство. Фагоцитоз апоптотических телец макрофагами или другими клетками активируется рецепторами на этих клетках: они захватывают и поглощают апоптотические клетки. Один из таких рецепторов на макрофагах – рецептор витронектина, который является β3-интегрином и активирует фагоцитоз апоптотических нейтрофилов.
Участие апоптоза в физиологических и патологических процессах
Запрограммированном разрушении клеток во время эмбриогенеза (включая имплантацию, органогенез). Несмотря на то, что при эмбриогенезе апоптоз не всегда является отражением “запрограммированной смерти клетки”, это определение апоптоза широко используют различные исследователи.
Гормон-зависимой инволюции органов у взрослых, например, отторжение эндометрия во время менструального цикла, атрезии фолликулов в яичниках в менопаузе и регрессия молочной железы после прекращения лактации.
Удалении некоторых клеток при пролиферации клеточной популяции.
Гибели отдельных клеток в опухолях, в основном при ее регрессии, но также и в активно растущей опухоли.
Гибели клеток иммунной системы, как В -, так и Т-лимфоцитов, после истощения запасов цитокинов, а также гибели аутореактивных Т-клеток при развитии в тимусе.
Патологической атрофии гормон-зависимых органов, например, атрофии предстательной железы после кастрации и истощении лимфоцитов в тимусе при терапии глюкокортикоидами.
Патологической атрофии паренхиматозных органов после обтурации выводных протоков, что наблюдается в поджелудочной и слюнных железах, почках.
Гибели клеток, вызванных действием цитотоксических Т-клеток, например, при отторжении трансплантата и болезни “трансплантат против хозяина”.
Повреждении клеток при некоторых вирусных заболеваниях, например, при вирусном гепатите, когда фрагменты апоптотических клеток обнаруживаются в печени, как тельца Каунсильмана.
Гибели клеток при действии различных повреждающих факторов, которые способны вызвать некроз, но действующих в небольших дозах, например, при действии высокой температуры, ионизирующего излучения, противоопухолевых препаратов.
Биохимия апоптоза.
Активация цистеиновых (и некоторых других) протеаз — наиболее универсальная черта программируемой клеточной гибели независимо от организма, в котором она происходит. Основные участники программируемой клеточной гибели, каспазы («caspase» от «cysteine aspase») — это семейство эволюционно консервативных цистеиновых протеаз, которые специфически расщепляют белки по остаткам аспарагиновой кислоты. В настоящее время идентифицировано 10 каспаз. При апоптозе помимо активации цистеиновых протеаз, у растений выявлено возрастание активности сериновой и аспарагиновой протеаз.
Кроме того, в апоптозе принимают участие и другие протеазы, прежде всего, кальпаины, или Са2+-зависимые протеазы и убиквитин (протеаза, ковалентно связывающаяся с белком-мишенью). Эти протеазы — обязательный компонент каскада протеолитических ферментов. Так, ингибиторы кальпаина блокируют апоптоз. Убиквитин-протеосомный путь деградации белков активируется при апоптозе.
Роль каспаз в апоптозе разнообразна. Результатом активности протеаз являются характерные изменения в морфологии клеток при апоптозе.1. Гидролиз белков ламинов, армирующих ядерную мембрану. Это ведет к распаду ядерной оболочки и конденсации хроматина. Мишенями протеаз при апоптозе являются также белки ядрышек, гистоны и негистоновые белки и топоизомераза. Топоизомераза — связующее звено между ДНК хроматина и белковыми структурами ядра, с помощью которого хроматин прикрепляется к ядерному матриксу. Расщепление топоизомеразы — это этап образования высокомолекулярных фрагментов ДНК.
2. Расщепление антиапоптозных белков — протеолиз ингибитора ДНКазы, ответственной за фрагментацию ДНК. В нормальных клетках апоптозная ДНКаза CAD (caspase-activated DNase) образует неактивный комплекс с ингибитором 1CMiwm DFF (DNA fragmentation factor). При апоптозе ингибитор Гмс участием каспаз 3 и 7 инактивируется и свободная CAD, вызывая нуклеосомные разрывы хроматина, ведет к образованию фрагментов ДНК с молекулярной массой кратной молекулярной массе ДНК в нуклеосомных частицах — 180-200 пар нуклеотидов. Эти фрагменты и дают характерную лесенку ДНК при электрофоретическом разделении в агарозном геле. Апоптоз возможен и без фрагментации ДНК. Обнаружен ядерный белок ACCINVS (apoptotic chromatin condensation inducer in the nucleus), который при комбинированном действии каспазы 3 и неидентифицированной протеазы расщепляется на фрагменты. Один из них в присутствии дополнительных неядерных факторов вызывает апоптотическую конденсацию хроматина и фрагментацию ядра (кариорексис) без фрагментации ДНК. Кроме непосредственной активации нуклеаз, протеазы (путем ограниченного протеолиза) устраняют структурное разобщение между нуклеазами и ДНК в составе хроматина, удаляют белки, защищающие ДНК.3. Угнетение репарации ДНК: инактивирование и нарушение регуляции белка, участвующего в репарации ДНК, а также в сплайсинге мРНК, репликации ДНК. Мишенью каспаз является поли-(АДФ-рибозо)-полимераза (ПАРП), которая участвует в репарации ДНК (катализирует полиАДФ-рибозилирование белков, связанных с ДНК). Донором АДФ-рибозы является NAD'. Активность ПАРП-полимеразы возрастает в 500 раз и более при связывании с участками разрыва ДНК. ПАРП участвует в репарации поврежденной ДНК, регуляции активности эндонуклеаз, поддержании структуры хроматина посредством АДФ-рибозилирования. Апоптотическая гибель клетки сопровождается расщеплением ПАРП каспазами. При массированных разрывах ДНК чрезмерная активация ПАРП, сильно снижая содержание внутриклеточного NAD*, ведет к подавлению гликолиза и митохондриального дыхания и вызывает гибель клетки по пути некроза.4. Разрушение белков цитоскелета. Деградация структурных и функциональных белков митотического аппарата.5. Участие в экспрессии генов. Эта функция связана с протеолизом репрессоров и с образованием пептидов, регулирующих транскрипцию (модификация факторов транскрипции). Субстратом протеаз является, например, гистон, выступающий репрессором генов.6. Одна из функций протеаз — передача апоптозного сигнала от индукторов апоптоза. Сигналы могут быть трансмембранными, рецептор-зависимыми. Рецепторами служат трансмембранные белки. Протеазы принимают участие либо непосредственно при взаимодействии индукторов апоптоза с рецепторами, либо через активацию протеинкиназ, играющих важную роль в передаче трансмембранного сигнала с целого ряда рецепторов.Локализация протеаз в различных отделах (компартментах) клетки способствует эффективной трансмембранной передаче сигналов программируемой клеточной гибели. Часть протеаз связаны с мембранами (цитоплазматической, ядерной, мембранами органелл или вакуоли) — это мембраносвязанные протеазы. Другие — находятся в матриксе ядра, цитоплазмы или органелл. Аспарагиновая протеаза растений, по всей видимости, локализована в вакуоли. Предполагается, что сериновые протеазы локализуются в цитоплазме и в ядре. Известно, что в ядрах протеазы могут быть прочно ассоциированы с хроматином и, в том числе, непосредственнно с гистонами. Перемещение протеаз в клетке может сопровождаться их активацией. Например, повышение концентрации Ca2+ внутри клетки способствует перемещению Са2+-зависимой протеазы и протеинкиназы из цитоплазмы в мембрану. При этом происходит автокаталитическая активация неактивных форм протеазы.Так, активация некоторых протеаз может быть обусловлена увеличением концентрации кальция в клетках, наблюдаемой при разных типах апоптоза (раздел выше). АФК также могут быть непосредственными индукторами активации протеаз. Появление локальных участков однонитевой ДНК активирует, например, ядерные ДНК-зависимые сериновые протеазы, специфичные к гистону.
Множество ветвей сигнальной трансдукции перепроверяет правильность выбранного алгоритма событий на пути к апоптозу, уберегая клетку от бессмысленной гибели. Выявлено несколько механизмов, ограждающих клетку от случайного самоуничтожения с участием протеаз.
Во-первых, протеазы синтезируются в клетке в неактивной форме, а процессинг неактивных форм протеаз происходит путем автолиза или путем протеолиза другими протеазами. Например, каспазы синтезируются в клетке в виде прокаспазы — неактивного мономера с молекулярной массой 30-50 кДа. Активные формы — тетрамеры, содержащие по две субъединицы: (р 10 — р20)2 (рис. 9.7). Прокаспазы обладают незначительной протеолитической активностью, составляющей 1-2% активности зрелой каспазы. Механизм протеолитического само- или перекрестного расщепления (ауто- или транс-процессинга), а затем пространственного сближения ведет к образованию активных каспаз. От прокаспазы отделяется регуляторный N-концевой домен (продомен), а оставшаяся часть молекулы разделяется на большую (около 20 кДа) и малую (около 10 кДа) субъединицы. Затем происходит ассоциация большой и малой субъединиц. Два гетеродимера образуют тетрамер с двумя каталитическими центрами, работающими независимо. Первоначально концентрация каспаз в клетке ничтожна. Благодаря свойству автокатализа, концентрация активных каспаз может возрастать лавинообразно.Во-вторых, протеазы обратимо взаимодействуют с эндогенными белковыми ингибиторами, образуя неактивные комплексы (латентные комплексы описаны для цистеиновых, Са2+-зависимых и некоторых других протеаз). При действии различных индукторов апоптоза происходит диссоциация неактивных комплексов протеаза-ингибитор. Обратимое взаимодействие Са2+-зависимых протеаз с эндогенными ингибиторами регулируется кальцием. Цистеиновая протеаза связывается ковалентно с ингибитором через дисульфидную связь. Высвобождение и активация каспазы происходит в результате тиол-дисульфидного обмена и сопряжена с окислительно-восстановительным состоянием клетки и метаболизмом глюкозы.В-третьих, протеазы могут быть компонентами специальных рецептор-зависимых систем. Так, [рецептор + лиганд + адаптер + прокаспаза] формируют специфический агрегат, в котором происходит активация каспаз. Такой агрегат называют апоптосомой или апоптозным шапероном. Самое интересное, что выявлены консервативные области гомологии (в том числе NB-область) белка адаптера у животных и продуктов генов резистентности у растений, включая томат, арабидопсис и табак. Более того, белки похожи структурно. Предполагается, что продукты гена резистентности могут играть роль адаптеров в апоптосоме. Таким образом, при узнавании продукта авирулентности, по всей видимости, происходит диссоциация апоптосомы и развертывание программы апоптоза.
Продукты генов резистентности, по-видимому, ответственны за эффективность гибели клеток при заражении — узнавание факторов и запуск машины самоуничтожения, за первые (самые важные) шаги на пути к стремительной гибели клетки.Существует несколько путей реализации программы ПКГ. Путь передачи сигнала: индукторы — рецепторы — адаптеры — каспазы первого эшелона — регуляторы — каспазы второго эшелона. Рецептор взаимодействует с лигандом. Насколько обратима гибель клетки? На этапе активации каспаз первого эшелона жизнь клетки еще можно сохранить. Существуют регуляторы, которые блокируют или, напротив, усиливают разрушительное действие каспаз первого эшелона. После активации каспазами первого эшелона каспаз второго эшелона путем протеолиза из прокаспаз процесс, запушенный программой смерти, становится необратим. Эти каспазы способны в дальнейшем к самоактивации (автокатализу или автопроцессингу) и активируют фактор фрагментации ДНК на нуклеосомные фрагменты. Вернемся к митохондриям. Апоптотическое изменение митохондрии может индуцироваться окислительным стрессом, повышением концентрации Ca2+. При апоптозе из межмембранного пространства митохондрий высвобождаются белки — апоптогенные факторы:
AIF (Apoptosis Inducing Factor) — флавопротеин с молекулярной массой 57 кДа. Будучи добавлен к изолированным ядрам, он вызывает конденсацию хроматина и фрагментацию ДНК, а при добавлении к изолированным митохондриям — высвобождение цитохрома С и каспазы 9. Высвобождаемый цитохром С вместе с цитоплазматическим фактором APAF-1 (apoptosis protease activating factor-1) образует комплекс с прокаспазой. APAF-I играет роль арматуры, на которой происходит аугокаталитический процессинг каспазы 9 (мультимерная арматура APAF1-цитохром-С-комплексов напоминает пропеллер). Обнаружены ингибиторы высвобождения цитохрома С, блокирующие апоптоз, например, белок Bel.
Список используемой литературы:
Гордеева А.В., Лабас Ю.А., Звягильская Р.А.Апоптоз одноклеточных организмов: механизмы и эволюция Биохимия, 2004, том 69, вып. 10, с. 1301—1313
Голубев А.М., Москалева Е. Ю., Северин С.Е., Веснянко Т.П., Кузовлев А.Н., Алкадарский А.С., Порошенко Г.Г. Апоптоз при критических состояниях
Читайте также: