Синдром Смита-Магениса - этиология, клиника, диагностика
Обновлено: 04.05.2024
Синдром Смита-Лемли-Опитца - этиология, клиника, диагностика
Синдром Смита-Лемли-Опитца недавно стал объектом пристального внимания неврологов и психиатров из-за отчетливой взаимосвязи с аутистическими проявлениями. Возможно, данное заболевание имеет генетическую природу и тесно связано с аутистическими симптомами (см. ниже).
Синдром Смита-Лемли-Опитца (ССЛО) регистрируется с частотой около 1 на 70000 новорожденных. Характерна высокая перинатальная смертность и высокая частота мертворождений. Среди выживших детей не менее чем у 80% отмечаются тяжелые отклонения, а у оставшихся 20% изменения фенотипа выражены умеренно.
Синдром Смита-Лемли-Опитца (ССЛО) до сих пор диагностируется на основании типичных клинических признаков. Заболевание вызвано врожденным нарушением постскваленового синтеза холестерина. Нарушение синтеза холестерина при ССЛО вызвано наследственной мутацией гена 3-b-гидроксистерол-d-7 редуктазы (DHCR7). Повреждение гена DHCR7 приводит к нарушению синтеза холестерина и десмостерола, приводящее к повышению уровня 7DHC/8DHC, характерному снижению уровня холестерина и, что особенно важно, дисморфическому развитию.
Открытие синдрома Смита-Лемли-Опитца вызвало новые вопросы о роли путей биосинтеза холестерина в развитии человека. В настоящее время у 250 пациентов с синдромом Смита-Лемли-Опитца идентифицирована 121 мутация; разнообразие мутаций обеспечивает различную клиническую выраженность. Для воспроизведения некоторых аномалий развития, характерных для ССЛО, и выяснения патогенеза заболевания, было создано две генетических модели синдрома на мышах.
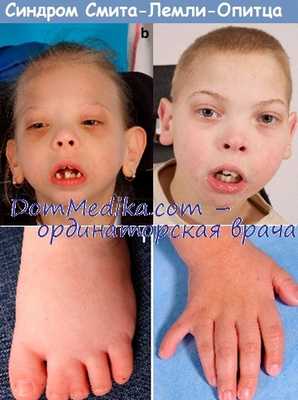
Наиболее характерными проявлениями синдрома наряду с задержкой умственного развития, аутизмом, другими поведенческими нарушениями и гипотонией является необычное строение лица с высоким плоским лбом, вывернутыми вперед ноздрями и микрогнатией, а также аномалии гениталий у мальчиков, включая крипторхизм и (или) гипоспадию, а в крайних случаях—неразвитые мужские гениталии, несмотря на нормальный кариотип XY (Scarbrough et al., 1986). Другие проявления включают аномальное строение ушных раковин, птоз век, эпикант, поперечное возвышение ладони и различные аномалии конечностей и внутренних органов (Opitz et al., 1994).
У некоторых пациентов отмечаются припадки, достаточно часто встречается отсутствие аппетита. В течение первого года жизни смертность высокая. Аномалии головного мозга могут включать микроцефалию, гипоплазию лобных долей, аномальное строение извилин и гипоплазию мозжечка. Случаи наиболее выраженного поражения иногда выделяются как ССЛО 2 типа (Curry et al., 1987).
Синдром наследуется аутосомно-рецессивным путем. Явное повышение частоты заболевания у мужчин, вероятно, связано с более легкой диагностикой на основании очевидных аномалий половых органов.
Концентрация холестерина в плазме очень низкая, в то время как содержание 7-гидрохолестерола повышено, что предполагает дефект фермента, приводящего к разрыву двойной связи гидрохолестерола (Tint et al., 1994). Выживаемость строго коррелирует с более высоким уровнем холестерина плазмы (Tint et al., 1995). Возможна пренатальная диагностика путем определения содержания 7-дегидрохолестерола в амниотической жидкости (Dallaire et al., 1995). Потребление большого количества холестерина может нормализовать его уровень в крови, но необязательно будет клинически эффективно (Ullrich et al., 1996).
Синдром Смит-Магенис
Синдром Смит-Магенис — это редкое генетическое заболевание, которое возникает при микроповреждении короткого плеча 17-й хромосомы. Патология проявляется множественным врожденными пороками развития, среди которых преобладают аномалии лицевого скелета, а также интеллектуальными нарушениями, разнообразными поведенческими особенностями. Для диагностики синдрома Смит-Магенис назначается молекулярно-генетическое тестирование, неврологический обследование, методы инструментальной визуализации. Больным требуется помощь логопедов, реабилитологов, коррекционных педагогов, по показаниям проводится лечение соматических осложнений.
МКБ-10
Общие сведения
Синдром назван в честь американских врачей ‒ клинициста Энн Смит и цитогенетика Эллен Магенис, которые в 1980 г. впервые описали характерный симптомокомплекс у группы детей. Ранее считалось, что заболевание встречается с частотой 1:25000, но с развитием молекулярно-генетических методов диагностики подтвердилась более высокая распространенность — 1:15000. Чаще всего патология наблюдается у детей с задержкой психического развития — 1 случай на 569 пациентов.
Причины
Заболевание возникает вследствие спонтанной хромосомной аномалии — делеции 17 хромосомы в локусе 17p11.2, которая предполагает потерю до 4 млн. нуклеотидных пар. До 25% клинических случаев синдрома Смит-Магенис составляют атипичные микроделеции, которые выражаются в утрате меньшего участка короткого плеча хромосомы. Болезнь носит спорадический характер, пока ученым не удалось установить типичные закономерности и провоцирующие факторы.
Патогенез
Развитие характерной для синдрома симптоматики прежде всего связывают с нарушением гена RAI1, который является индуктором ретиноловой кислоты. Ученые обсуждают его участие в дифференцировке тканей, что объясняет формирование множественных врожденных пороков. Также RAI1 предположительно используется при дифференцировке нейронов. Некоторые психические проявления синдрома Смит-Магенис вызваны дефектом синтеза и деградации меланина.
Симптомы
Дети имеют типичные особенности внешности, которые обнаруживаются уже в младенческом возрасте. Обычно у них широкая квадратная форма головы, выпуклый лоб, глубоко посаженные глаза с монголоидным разрезом. Также для синдрома характерно недоразвитие средней зоны лица, короткий нос с вывернутыми ноздрями, шатровидная верхняя губа при относительно маленькой нижней челюсти. У детей непропорционально короткие руки и ноги, с раннего возраста отмечается задержка роста.
Специфическим диагностическим признаком синдрома Смит-Магенис являются нарушения сна. На первом году жизни наблюдается патологическая сонливость, такие дети много спят днем, их приходится будить для очередного кормления. Со временем нарушаются циркадные ритмы, преобладает поздний отход ко сну с частыми ночными пробуждениям. С возрастом пациент все больше спит днем, не может заснуть ночью.
Психоневрологические изменения включают умственную отсталость умеренной степени тяжести, возникающую практически у всех больных. Характерна задержка психоречевого развития, затруднения при произношении слов, дефицит внимания с гиперактивностью. До 30% пациентов имеют неврологические нарушения: расстройства координации движений, периферическую нейропатию, судорожные приступы.
Болезнь Смит-Магенис сопровождается специфическим поведенческим профилем. Дети страдают стереотипным поведением, проявляют дезадаптивные черты личности — вспышки гнева, непослушание, постоянное требование внимания к себе со стороны взрослых. Некоторые больные склонны к самоповреждению: кусают, щиплют или бьют себя, вводят инородные предметы в физиологические отверстия тела.
Осложнения
Большинство детей имеют мышечную гипотонию с рождения, вследствие чего возникают трудности вскармливания, нарушения глотания, отказ от пищи. В 80% случаев синдром Смит-Магенис ассоциирован с офтальмологическими проблемами: дисплазией радужки, микрокорнеа, пятнами Брушфильда. Более 60% больных страдают хроническими отитами, кондуктивной или нейросенсорной тугоухостью. У половины пациентов диагностируется гиперхолестеринемия.
Пороки сердца встречаются в 25% случаев синдрома. Наиболее распространены дефекты межпредсердной и межжелудочковой перегородок, стеноз трикуспидального клапана, тетрада Фалло. До 15% детей страдают от аномалий мочевыводящих путей, которые включают унилатеральную агенезию почек, удвоение мочеточников, увеличение линейных размеров органов.
Диагностика
Обследование осуществляет мультидисциплинарная команда специалистов с участием педиатра, детского невролога, психиатра, генетика. Синдром Смит-Магенис имеет патогномоничные фенотипические и поведенческие признаки, при выявлении которых устанавливается предварительный диагноз. Для верификации патологии используются следующие методы:
- Генетический анализ. Для обнаружения мутации применяется FISH гибридизация со специфическими зондами, которая определяет патологию с точностью 96-100%. Если тест не показал синдром Смит-Магенис при наличии типичных клинических признаков, выполняются дополнительные молекулярные анализы.
- Пренатальная диагностика. При отягощенном семейном анамнезе или настораживающих результатах УЗИ для исключения синдрома возможно исследование путем амниоцентеза во втором триместре беременности. Учитывая спонтанный характер мутации, такая диагностика производится редко.
- Неврологический осмотр. Пациенту требуется полное обследование с оценкой рефлексов, мышечного тонуса, проб на координацию движений. Обязательно проводятся тесты на уровень интеллекта, степень развития когнитивных функций. Зачастую осмотр дополняется консультацией психиатра.
- Инструментальная визуализация. Методы исследования назначаются дифференцировано. Может потребоваться офтальмоскопия, аудиометрия, исследование вызванных слуховых потенциалов. Для выявления соматических нарушений, типичных для клинической картины синдрома, рекомендованы эхокардиография, УЗИ брюшной полости и забрюшинного пространства.
Лечение синдрома Смит-Магенис
Показано ежегодное комплексное обследование состояния здоровья для контроля динамики нарушений. Больному подбирается индивидуальная программа симптоматической терапии. Медикаментозные средства назначаются редко, в основном для коррекции бессонницы, а психотропные препараты не используются из-за риска усугубления психических проблем. Лечение включает следующие направления:
- Логопедическая помощь. В раннем возрасте занятия с логопедом необходимы для улучшения состояния артикуляционного аппарата и структур полости рта, что облегчает кормление, создает условия для развития речи. В дальнейшем требуется продолжительная коррекция для усовершенствования вербальных навыков.
- Эрготерапия. Частые проблемы с мышечным тонусом и координацией движений требуют участия в программе лечения реабилитологов, которые помогают восстановить моторные функции, развить мелкую моторику, усовершенствовать зрительную и слуховую перцепцию.
- Коррекционные программы. Дети с болезнью Смит-Магенис хорошо учатся в небольших тихих коллективах до 5-7 человек, где каждый получает достаточное внимание и поддержку от преподавателя. Учебные программы подбираются с учетом степени интеллектуальных нарушений.
При осложненном течении синдрома схема лечения расширяется. Неврологические расстройства требуют применения физиотерапии, упражнений ЛФК, противоэпилептических препаратов. Для нормализации зрения некоторым необходима оптическая коррекция, исправление косоглазия. Хирургическое лечение пороков сердца осуществляется в соответствии с выявленной аномалией.
Прогноз и профилактика
При своевременном начале комплексной терапии синдрома Смит-Магенис удается значительно развить моторные и речевые навыки пациента, адаптировать его к жизни в социуме. Прогноз относительно благоприятный при пожизненной реабилитации, социально-психологической поддержке. Специфические меры профилактики синдрома не разработаны, что обусловлено его спорадическим характером.
1. Smith-Magenis Syndrome / Smith, A. C., & Gropman, A. L. // Cassidy and Allanson's Management of Genetic Syndromes. — 2021.
2. Синдром Смит-Магенис: клиникогенетическая характеристика и молекулярно-цитогенетическая диагностика/ О.М. Хурс, Н.В. Румянцева, Л.В. Исакович, О.Л. Зобикова// Педиатрия. Восточная Европа. — 2015. — №1.
Синдром Меллори-Вейса ( Желудочно-пищеводный разрывно-геморрагический синдром )
Синдром Меллори-Вейса — линейные разрывы слизистой кардиоэзофагеальной зоны, возникшие на фоне рвоты, позывов на рвоту, икоты. Проявляется наличием крови в рвотных массах, эпигастральными или загрудинными болями, артериальной гипотензией, тахикардией. Диагностируется с помощью эзофагогастроскопии, обзорной рентгенографии брюшной полости. Для лечения применяется гемостатическая, кровезаместительная терапия, противорвотные препараты, сердечные аналептики, ингибиторы протонной помпы, Н2-гистаминоблокаторы, антациды. При необходимости выполняется эндоскопический гемостаз, терапевтическая эмболизация, гастротомия для ушивания повреждений.
Впервые клиника разрывно-геморрагического синдрома была описана в 1929 году американскими патологами Дж.К. Меллори и С. Вейсом. В настоящее время заболевание является одной из ведущих причин неязвенных кровотечений из верхних отделов пищеварительного тракта. Распространенность патологии достигает 5-10%. Болезнь Меллори-Вейса выявляется преимущественно у 45-60-летних пациентов, злоупотребляющих спиртными напитками. У мужчин разрывы желудочной и пищеводной слизистой возникают в 7 раз чаще, чем у женщин. У 79-80% больных поражается эзофагогастральный переход, у 16-17% — стенка пищевода, у 3-5% — кардиальная оболочка. Длина разрывов обычно составляет 0,4-4,5 см. В 77-78% случаев повреждения являются единичными, в 22-23% — множественными.
Продольные разрывы слизистой в области пищеводно-желудочного перехода возникают при локальном повышении давления у пациентов со сниженной резистентностью эпителиального слоя. Предпосылками к развитию разрывно-геморрагического гастроэзофагеального синдрома служат патологические процессы, при которых повреждаются эпителиоциты или наблюдается повышенное кровенаполнение сосудов верхних отдела ЖКТ: асептическое воспаление слизистой при частом употреблении спиртных напитков, воспалительные заболевания ЖКТ (эзофагиты, гастриты), длительный прием НПВС, кортикостероидов, скользящая грыжа пищеводного отверстия диафрагмы, расширение пищеводных вен при портальной гипертензии у больных с гепатитами, жировым гепатозом, фиброзом, циррозом печени. Непосредственными причинами болезни Меллори-Вейса являются:
- Рвота, неукротимая икота. У 80-85% пациентов развитие рвоты связано с алкогольным опьянением. Провоцирующими факторами также становятся рвота беременных, диспепсические расстройства при патологии пищеварительного тракта (язвенной болезни, панкреатите, холецистите), отравления, уремия.
- Длительный интенсивный кашель. В редких случаях разрывно-геморрагический синдром провоцируется острыми и хроническими респираторными заболеваниями. Линейное повреждение слизистой пищевода, верхних отделов желудка может осложнить коклюш, ОРВИ, хронический бронхит, бронхиальную астму.
- Ятрогенные воздействия. Повреждение стенки пищевода, желудка возможно при грубом выполнении эндоскопических манипуляций (гастроскопии, эзофагогастродуоденоскопии), введении желудочного зонда. Иногда разрывы слизистой возникают при проведении сердечно-легочной реанимации.
В спорадических случаях повышение давления, приводящее к разрыву эпителиального слоя, вызывается другими факторами — подъемом тяжестей, интенсивными физическими нагрузками с резким напряжением мышц брюшного пресса, тупой травмой живота. Крайне редко заболевание осложняет течение судорожного синдрома при эпилепсии, опухолях головного мозга, энцефалопатиях, менингите, энцефалите, эклампсии.
Пусковым моментом разрыва пищеводно-желудочной слизистой обычно становится многократная рвота, резкое повышение абдоминального давления при переполненном желудке или кардиоэзофагеальном спазме, реже — прямые механические воздействия. Возникновение избыточного давления в кардиальном отделе желудка способствует перерастяжению стенки органа. При морфологической несостоятельности эпителия, вызванной воспалительными процессами, растянутая слизистая желудка, пищевода разрывается в наиболее истонченном или патологически измененном участке. Обычно разрыв распространяется не глубже эпителиального и подслизистого слоя. В тяжелых случаях повреждается мышечная, серозная желудочная либо адвентициальная пищеводная оболочки с выходом агрессивного содержимого в средостение или брюшную полость.
Классификация
- I стадия. Повреждение слизистой желудочно-пищеводного перехода и дистальной трети пищевода. Встречается у 36-37% пациентов. В большинстве случаев кровотечение прекращается спонтанно.
- II стадия. Дефекты расположены в той же зоне, однако их глубина достигает подслизистого слоя. Выявляется у 52-53% больных. Обычно проводится консервативная гемостатическая терапия.
- III стадия. Глубокие зияющие разрывы с вовлечением мышечной оболочки и интенсивным кровотечением. Наблюдаются в 9-11% случаев. Необходим эндоскопический или хирургический гемостаз.
- IV стадия. Редко диагностируемое тяжелое повреждение с разрушением всех оболочек гастроэзофагеального участка ЖКТ. Осложняется медиастинитом, перитонитом, пневмотораксом.
Симптомы синдрома Меллори-Вейса
Клинические проявления заболевания обычно развиваются на фоне многократной рвоты. Основным признаком синдрома является выделение ярко-красной крови с рвотными массами (гематемезис), которое может иметь различную интенсивность – от нескольких капель до профузного кровотечения. Возникает резкая боль в эпигастральной области или за грудиной. Вследствие кровопотери у больного формируется острый анемический синдром, для которого характерны головокружение, бледность кожных покровов, мелькание «мушек» перед глазами, падение артериального давления, значительное учащение сердцебиения. При массивном кровотечении возможна потеря сознания.
Острая кровопотеря при симптомокомплексе Меллори-Вейса может привести к развитию геморрагического шока с тяжелыми нарушениями микроциркуляции, изменениями реологических свойств крови, прогрессирующей гипоксией. При отсутствии лечения шок переходит в декомпенсированную стадию, сопровождающуюся полиорганной недостаточностью. Наиболее тяжелым осложнением синдрома является тотальный разрыв стенки брюшного отдела пищевода, распространяющийся выше уровня диафрагмы. При этом у пациента возникает приступ одышки, цианоз кожи, сильнейшие боли в грудной клетке. Такое осложнение, известное как синдром Бурхаве, в 20-40% случаев заканчивается летальным исходом. Попадание содержимого желудка в средостение, полость брюшины провоцирует развитие медиастинита, перитонита.
Постановка диагноза при синдроме Мэллори-Вейса может быть затруднена, что обусловлено стремительным нарастанием клинической картины и необходимостью оказания пациенту экстренной медицинской помощи. Диагностика заболевания предполагает комплексное инструментальное обследование пищеварительного тракта для выявления первопричины кровавой рвоты. Наиболее информативными являются:
- Эзофагогастроскопия. Введение гибкого эндоскопа через ротовую полость позволяет оценить состояние эпителиальной оболочки верхних отделов ЖКТ и обнаружить линейные разрывы, которые обычно локализованы в области перехода пищевода в желудок. С помощью визуального осмотра удается установить глубину поражения стенки пищевода или желудка.
- Обзорная рентгенография брюшной полости. Проведение рентгенологического исследования информативно при подозрении на разрыв полого органа. Основной признак перфорации – наличие свободного газа в полости брюшины (симптом «серпа»). На рентгенограмме также можно обнаружить другие болезни ЖКТ, которые являются первопричиной патологии Меллори-Вейса.
В клиническом анализе крови определяются изменения, характерные для анемического синдрома — уменьшения содержания эритроцитов и гемоглобина, снижение показателя гематокрита. Для исключения хронического кишечного кровотечения проводится реакция Грегерсена, позволяющая обнаружить скрытую кровь в кале. При выраженном диспепсическом синдроме может выполняться бактериологический посев кала для выявления патогенных микроорганизмов.
Дифференциальная диагностика синдрома осуществляется с легочным кровотечением, отеком легких, сердечной астмой, кровотечением из язвы желудка, варикозным расширением пищеводных вен, острым гастроэнтеритом, кишечными инфекциями, распадом опухоли желудка или пищевода, синдромом Рандю-Ослера. Кроме осмотра хирурга и гастроэнтеролога пациенту могут потребоваться консультации гематолога, инфекциониста, пульмонолога, кардиолога, гематолога, гепатолога.
Лечение синдрома Меллори-Вейса
Пациент подлежит неотложной госпитализации в хирургический стационар. На начальном этапе больному обеспечивается покой, холод на область желудка, при позывах на рвоту применяются блокаторы дофаминовых и серотониновых рецепторов с противорвотным эффектом. Назначается консервативное лечение и малоинвазивные манипуляции, направленные на остановку кровотечения, восполнение объема циркулирующей крови. При резком падении АД терапию дополняют введением средств для поддержания гемодинамики. Пациентам с болезнью Меллори-Вейса показаны:
- Инфузионная терапия. При умеренной кровопотере проводятся внутривенные вливания коллоидных и кристаллоидных растворов. При массивном кровотечении переливается эритроцитарная масса или взвесь, нативная и свежезамороженная плазма, реже — донорская кровь.
- Гемостатические препараты. Для медикаментозного гемостаза используют стимуляторы свертывающей системы крови. Эффективность кровоостанавливающей терапии повышается при парентеральном введении препаратов кальция, синтетических аналогов витамина К.
- Эндоскопический гемостаз. При продолжающемся кровотечении с помощью эндоскопа обкалывают место повреждения средствами с сосудосуживающим эффектом, вводят склерозанты, лигируют или клипируют сосуды. Возможно выполнение аргоноплазменной или электрокоагуляции.
- Терапевтическая эмболизация. Для прекращения кровотечения из поврежденных сосудов в них под контролем ангиографии вводят смесь эмболов с физиологическим раствором. Альтернативным методом является редко применяемое внутриартериальное вливание жировых суспензий.
Баллонная зондовая тампонада используется ограниченно из-за возможного усугубления разрывов. Важным условием быстрого восстановления поврежденной стенки является угнетение желудочной секреции при помощи ингибиторов протонной помпы, блокаторов Н2-гистаминорецепторов. Прием секретолитиков дополняют назначением невсасывающихся антацидов, препаратов коллоидного висмута. Хирургические методы лечения геморрагического разрывного синдрома показаны при неостанавливающихся или рецидивирующих кровотечениях, глубоких дефектах, полном разрыве пищеводной или желудочной стенки. Рекомендованным вмешательством является гастротомия с прошиванием надрывов, кровоточащих сосудов, ушиванием дефектов, иногда — перевязкой левой желудочной артерии.
Исход патологического состояния зависит от величины кровопотери и тяжести основного заболевания пациента. В 90% случаев кровотечение останавливается самопроизвольно или консервативными способами. Прогноз синдрома относительно неблагоприятный при потере больше 10% ОЦК и наличии сопутствующей патологии. Меры профилактики при заболевании Меллори-Вейса заключаются в отказе от злоупотребления алкоголем, своевременном устранении провоцирующих факторов, выявлении и лечении болезней желудочно-кишечного тракта, соблюдении техники проведения инвазивных медицинских манипуляций на пищеводе, желудке.
2. Лечение разрывно-геморрагического синдрома (синдрома Маллори-Вейсса) в специализированном Центре: автореферат диссертации/ Чередников Е.Е. – 2011.
Синдром Смита-Лемли-Опица
Синдром Смита-Лемли-Опица — это редкое наследственное заболевание, которое связано с генетическим дефектом обмена холестерина. Возникает при мутации гена DHCR7 в 11-й хромосоме, в локусе 11q12-q13. Патология проявляется низкорослостью, умственной отсталостью, многочисленными костными аномалиями и врожденными пороками внутренних органов. Диагностика синдрома предполагает биохимические анализы, генетическое тестирование, инструментальные способы визуализации соответственно превалирующей симптоматике. Лечение поддерживающее: нейрореабилитационные мероприятия, помощь пластических хирургов, кардиохирургов, челюстно-лицевых хирургов.
Синдром назван в честь американских педиатров Д. Смита, Л. Лемли и генетика Дж. Опица, которые в 1964 г. описали его клинические признаки. Генетические механизмы развития заболевания были установлены в 1998 г. Частота встречаемости синдрома составляет в среднем 1:29000, чаще болеют европейцы (1:22000). При этом носительство мутантного гена широко распространено: до 1,2% у светлокожего населения и до 0,8% у темнокожих людей. Состояние характеризуется тяжелым течением, частой инвалидизацией, высокой смертностью, что обуславливает его актуальность в современной генетике.
Синдром Смита-Лемли-Опица связан с аномалией (миссенс- и нонсенс-мутацией) гена DHCR7, который располагается на длинном плече 11 хромосомы (11q12-q13). Он кодирует фермент 7-дегидрохолестерин-редуктазу, участвующий в заключительном этапе синтеза холестерина. Заболевание имеет аутосомно-рецессивный тип наследования. Риск возникновения синдрома у ребенка, оба родителя которого являются носителями поврежденного гена, составляет 25%.
Основным механизмом, который провоцирует появление патогномоничных фенотипических признаков синдрома, считается холестериновый дефицит. При этом заболевании конечная молекула не образуется, вместо нее в организме накапливаются липопротеины низкой плотности, другие промежуточные метаболиты. Эти вещества в избыточных количествах обладают токсическим влиянием на клетки.
Неврологические нарушения вызваны недостаточным образованием липидов, которые необходимы для миелинизации нервных волокон, а специфический сигнальный белок sonic hedgehog провоцирует нарушения внутриутробного формирования мозговой ткани. Дефицит холестерина коррелирует с недостатком гормонов (тестостерона, эстрогенов), что объясняет нарушения формирования наружных половых органов у мальчиков.
Для болезни Смита-Лемли-Опица специфичны микроцефалия, волчья пасть, микрогнатия. Реже встречается синдром Пьера-Робена в сочетании с выступающими резцами, что создает затруднения при жевании пищи, проведении анестезиологического пособия. У 95% детей наблюдается Y-образная синдактилия 2-3 пальцев ног, в половине случаев отмечается постаксиальная синдактилия. Также характерны вывих тазобедренных суставов, укорочение конечностей.
При генетическом дефекте DHCR7 типично отставание в росте, физическом развитии. С самого рождения младенцы плохо набирают вес, длина их тела ниже нормы. По мере взросления ребенка задержка роста становится более заметной, во взрослом возрасте параметры тела находятся ниже 3-го перцентиля. Нарушается половое созревание: у мальчиков выявляются гипоспадия, крипторхизм, у девочек — гипертрофия клитора.
У большинства пациентов определяется выраженная неврологическая симптоматика. Она проявляется нарушениями сна (70% больных): частыми ночными пробуждениями, неспособностью уснуть, отсутствием четкого режима дня. Психические симптомы включают повышенную агрессивность, в том числе аутоагрессию, склонность к навязчивому поведению, расстройства аутистического спектра. Большинство детей, страдающих синдромом Смита-Лемли-Опица, умственно отсталые.
36-38% случаев синдрома осложняется врожденными сердечными пороками. Аномалии желудочно-кишечного тракта встречаются у 25-29% больных. Они включают гастроэзофагеальный рефлюкс, болезнь Гиршпрунга, мальротацию кишечника. Распространены пороки мочеполовой системы: кистозная дисплазия почек (29%), дистопия почек (19%), гидронефроз (16%) и обструкция лоханочно-мочеточникового соединения (13%). Около 12-18% пациентов страдают от снижения зрения.
Большинству больных устанавливается инвалидность из-за выраженной умственной отсталости, отставания темпов физического развития. Синдром Смита-Лемли-Опица ассоциирован с высоким уровнем смертности как в перинатальном периоде, так и после рождения ребенка вследствие грубых врожденных аномалий. Хотя многие пациенты доживают до зрелого возраста, им требуется постоянный уход и медицинское наблюдение.
При физикальном осмотре ребенка удается выявить патогномоничные изменения фенотипа, что вместе с признаками задержки роста и психомоторного развития позволяет поставить предварительный диагноз. Для подтверждения синдрома Смита-Лемли-Опица назначается обследование у генетика, ряд диагностических мероприятий:
- Биохимический анализ. В плазме крови и тканях организма проверяется содержание липидов. При этом генетическом заболевании наблюдается повышение уровня 7-дегидорохолестерина одновременно со снижением количества холестерина.
- Генетическое исследование. Для обнаружения специфической мутации выполняется секвенирование экзона. При выявлении аномалии DHCR7 диагноз синдрома Смита-Лемли-Опица подтверждается со 100% точностью. Такая же диагностика показана родственникам больного.
- Пренатальная диагностика. Парам с отягощенной наследственностью рекомендуется исследование во время беременности путем измерения концентрации 7-дегидрохолестерола, генетического анализа амниотической жидкости или ворсин хориона.
- Инструментальные методы. Пациентам проводится комплексное обследование сердечно-сосудистой (эхокардиография, ЭКГ), мочеполовой (экскреторная урография, УЗИ органов малого таза), нервной системы (ЭЭГ, нейросонография, КТ или МРТ головного мозга).
Лечение синдрома Смита-Лемли-Опица
Консервативная терапия
При наличии синдрома Смита-Лемли-Опица проводится симптоматическая терапия, которая подбирается индивидуально на основании ведущих клинических признаков. Пациентам требуется помощь мультидисциплинарной команды врачей, поскольку у одного больного сочетаются разнообразные поражения внутренних органов, нарушения костно-мышечной системы, нервно-психические расстройства. Выделяют следующие направления лечения генетического синдрома:
- Диетотерапия. Для нормализации липидного профиля плазмы крови применяются пищевые добавки с кристаллическим холестерином, рацион обогащается яичными желтками, животными жирами.
- Реабилитация. Для улучшения моторных функций назначается комплексная программа ЛФК, массаж, кинезитерапия. Чтобы скорректировать нервно-психический дефицит, показаны длительные занятия с коррекционными педагогами, логопедами, психологами.
- Квалифицированный уход. Ввиду умственной отсталости пациенты не могут проживать самостоятельно. Уход за ними осуществляют родители с участием медицинского персонала, также рассматривается вариант пребывания больных в специализированных интернатах.
- Диспансерное наблюдение. Пациенты с синдромом Смита-Лемли-Опица должны регулярно проходить медицинские осмотры у ортопеда-травматолога, невролога, уролога и других специалистов. Частота обследований определяется тяжестью клинических симптомов.
Хирургическое лечение
Для устранения аномалий лицевого черепа, синдактилии, гипоспадии проводятся пластические и челюстно-лицевые вмешательства. Объем и сроки операции подбираются с учетом степени тяжести фенотипических изменений. Коррекция врожденных пороков сердца выполняется по стандартным протоколам в зависимости от характера аномалии, наличия расстройств кровообращения. Сложные пороки требуют вмешательства кардиохирургов уже на первом году жизни ребенка.
При своевременной диагностике синдрома и раннем начале комплексной терапии прогноз относительно благоприятный. Врачам удается ускорить психомоторное развитие, устранить аномалии строения скелета, скорректировать соматические патологии. Однако низкий рост, умственная отсталость, проблемы с фертильностью остаются на всю жизнь. Профилактика предполагает генетическое консультирование носителей мутировавшего гена перед планированием беременности.
1. Федеральные клинические рекомендации по диагностике и лечению синдрома Смита-Лемли-Опица/ Л.П. Назаренко. — 2015.
2. Врожденные пороки развития. Синдром Смита-Лемли-Опица/ О.А. Милованова, Н.В. Чернышева, А.И. Чубарова, Л.В. Калинина// Неврологический журнал. — 2013. — №3.
3. Случай синдрома Смита-Лемли-Опица/ Л.И. Ширяева, А.М. Поздняков, Е.А. Соляник// Научно-медицинский вестник Центрального Черноземья. — 2006. — №24.
4. Синдром Смита-Лемли-Опица у детей/ А.Н. Семячкина, П.В. Новиков, М.И. Яблонская, М.Б. Кубатов// Российский вестник перинатологии и педиатрии. — 2006. — №3.
Синдром Смита-Магениса. Это редкое генетическое заболевание, возникающее при микроповреждении короткого плеча 17-й хромосомы. Патология проявляется в нескольких врожденных пороках развития, среди которых преобладают аномалии лицевого скелета, а также интеллектуальные нарушения и различные особенности поведения. Для диагностики синдрома Смита-Магениса назначаются молекулярно-генетические тесты, неврологические обследования и инструментальные визуализационные тесты. Пациентам требуется помощь логопедов, реабилитологов, корректоров, соматические осложнения проходят лечение в зависимости от показаний.
Синдром Смит-Магенис
Дополнительные факты
Синдром назван в честь американских врачей - клинициста Энн Смит и цитогенетика Эллен Магенис, которые в 1980 году впервые описали характерный симптомокомплекс у группы детей. Ранее считалось, что заболевание возникает с частотой 1: 25000, но с развитием методов молекулярно-генетической диагностики подтверждена более высокая распространенность - 1: 15000. Чаще всего патология наблюдается у детей с задержкой умственного развития - 1 случай на 569 больных.
Заболевание возникает из-за спонтанной хромосомной аномалии - делеции 17 хромосомы в локусе 17p11.2, что предполагает потерю до 4 миллионов пар оснований. До 25% клинических случаев синдрома Смита-Магениса представляют собой атипичные микроделеции, которые выражаются в потере меньшей части короткого плеча хромосомы. Заболевание носит спорадический характер, пока ученым не удалось установить типичные закономерности и провоцирующие факторы.
Развитие симптомов, характерных для синдрома, связано, прежде всего, с нарушением гена RAI1, который является индуктором ретиноловой кислоты. Ученые обсуждают его роль в дифференцировке тканей, которая объясняет формирование многих врожденных дефектов. RAI1 также, возможно, используется для дифференцировки нейронов. Некоторые психологические симптомы синдрома Смита-Магениса вызваны нарушением синтеза и деградации меланина.
Клиническая картина
Дети обладают типичными физическими характеристиками, присущими младенческому возрасту. У них обычно широкая квадратная форма головы, выпуклый лоб, глубоко посаженные глаза с монголоидным вырезом. Для синдрома также характерны недоразвитие средней зоны лица, короткий нос с закрученными ноздрями, шатровидная верхняя губа с относительно небольшой нижней челюстью. У детей непропорционально короткие руки и ноги, и с раннего возраста отмечается задержка роста.
Нарушения сна являются специфическим диагностическим признаком синдрома Смита-Магениса. В первый год жизни наблюдается патологическая сонливость, такие дети много спят днем, их нужно разбудить для следующего кормления. Со временем нарушаются циркадные ритмы, преобладает поздний отход ко сну с частыми ночными пробуждениями. С возрастом пациент спит все больше днем и не может заснуть ночью.
Психоневрологические изменения включают умственную отсталость средней степени тяжести, которая встречается практически у всех пациентов. Характеризуется задержкой психоречевого развития, затруднением произношения слов, синдромом дефицита внимания с гиперактивностью. До 30% пациентов имеют неврологические расстройства: нарушения координации движений, периферическую невропатию, судороги.
Болезнь Смита-Магениса имеет специфический поведенческий профиль. Дети страдают стереотипным поведением, проявляют несоответствующие черты личности - вспышки гнева, непослушания, постоянные требования внимания со стороны взрослых. Некоторые пациенты склонны к членовредительству: кусают, щипают или дерутся, вставляют посторонние предметы в физиологические отверстия тела.
Ассоциированные симптомы: Судороги.
Возможные осложнения
Большинство детей страдают от низкого кровяного давления с рождения, что приводит к затруднениям с кормлением, проблемам с глотанием и отказу от еды. В 80% случаев синдром Смита-Магениса связан с офтальмологическими проблемами: дисплазией радужной оболочки, микрокорнеей, пятнами Брашфилда. Более 60% пациентов страдают хроническим средним отитом, кондуктивной или нейросенсорной тугоухостью. У половины пациентов диагностирована гиперхолестеринемия.
Пороки сердца встречаются в 25% случаев синдрома. Наиболее часто встречаются пороки предсердной и межжелудочковой перегородок, стеноз трехстворчатого клапана, тетралогия Фалло. До 15% детей имеют аномалии мочевыводящих путей, которые включают одностороннюю агенезию почек, удвоение мочеточников и увеличение линейных размеров органов.
Тест проводится многопрофильной командой специалистов с участием педиатра, детского невролога, психиатра и генетика. Синдром Смита-Магениса имеет патогномоничные фенотипические и поведенческие симптомы, при обнаружении которых ставится предварительный диагноз. Для верификации патологии используются следующие методы:
• Генетический анализ. Для выявления мутаций используется FISH-гибридизация со специфическими зондами, определяющая патологию с точностью 96-100%. Если тест не выявляет синдром Смита-Магениса при наличии типичных клинических симптомов, проводятся дополнительные молекулярные тесты.
• Пренатальная диагностика. При отягощенном семейном анамнезе или тревожных результатах УЗИ можно пройти амниоцентез во втором триместре беременности, чтобы исключить синдром. Учитывая спонтанный характер мутации, этот диагноз ставится редко.
• Неврологическое обследование. Пациенту требуется комплексное обследование с оценкой рефлексов, мышечного тонуса, пробы на координацию движений. Тесты на уровень интеллекта, степень развития когнитивных функций обязательны. Часто обследование дополняется консультацией психиатра.
• Инструментальный дисплей. Методы исследования назначаются дифференцированно. Может потребоваться офтальмоскопия, аудиометрия и исследование слуховых вызванных потенциалов. Для выявления соматических нарушений, характерных для клинической картины синдрома, рекомендуется эхокардиография, УЗИ брюшной полости и забрюшинного пространства.
Лечение
Было показано, что ежегодная комплексная проверка здоровья позволяет контролировать динамику заболеваний. Пациенту подбирается индивидуальная программа симптоматической терапии. Лекарства назначаются редко, в основном для коррекции бессонницы, а психотропные препараты не используются из-за риска обострения психических расстройств. Лечение включает следующие направления:
• Логопедическая помощь. С раннего возраста занятия с логопедом необходимы для улучшения состояния артикуляционного аппарата и структур полости рта, что облегчает прием пищи, создает условия для развития речи. В дальнейшем необходима длительная коррекция для улучшения речевых навыков.
• Трудотерапия. Распространенные проблемы с мышечным тонусом и координацией движений требуют участия в лечебной программе реабилитологов, которые помогают восстановить двигательные функции, развить мелкую моторику, улучшить зрительное и слуховое восприятие.
• Коррекционные программы. Дети с болезнью Смита-Магениса хорошо учатся в небольших тихих группах не более 5-7 человек, где каждой уделяется достаточно внимания и поддержки со стороны учителя. Программы подбираются с учетом степени умственной отсталости.
При осложненном течении синдрома режим лечения расширяется. Неврологические расстройства требуют применения физиотерапии, ЛФК, противоэпилептических препаратов. Для нормализации зрения некоторым нужна оптическая коррекция, коррекция косоглазия. Хирургическое лечение пороков сердца проводится в соответствии с выявленной патологией.
Прогноз
При своевременном начале комплексной терапии синдрома Смита-Магениса можно значительно развить двигательные и языковые навыки пациента, адаптировать его к жизни в обществе. Прогноз относительно благоприятный при постоянной реабилитации, социальной и психологической поддержке. Специфических мер по профилактике синдрома не разработано из-за его спорадического характера.
Список литературы
1. Smith-Magenis Syndrome / Smith, A. C., & Gropman, A. L. // Cassidy and Allanson s Management of Genetic Syndromes. — 2021.
2. Синдром Смит-Магенис: клиникогенетическая характеристика и молекулярно-цитогенетическая диагностика/ О.М. Хурс, Н.В. Румянцева, Л.В. Исакович, О.Л. Зобикова// Педиатрия. Восточная Европа. — 2015. — №1.
3. Федеральные клинические рекомендации по диагностике и лечению синдрома Смит-Магенис. — 2015.
Читайте также:
- Синоназальный аденокистозный рак - лучевая диагностика
- Идиопатическое негранулематозное воспаление глазницы (воспалительный псевдотумор, синдром идиопатического воспаления глазницы): признаки, гистология, лечение, прогноз
- Показания для аблации сердца и ее осложнения
- Невус белый беспигментный
- Классификация хронического гастрита в таблице