Синдром нечувствительности к андрогенам. Причины и диагностика
Обновлено: 25.04.2024
Андрогенный дефицит – это недостаточность тестостерона (мужского полового гормона) в организме мужчины. В подростковом возрасте проявляется задержкой полового созревания и отсутствием вторичных половых признаков. В зрелом возрасте вызывает снижение полового влечения, эрекции, уменьшение роста волос в андрогенозависимых зонах. Приводит к расстройству сексуальной жизни, бесплодию, личностным психологическим травмам. Диагностируется по данным гормональных тестов. Лечение подразумевает назначение заместительной гормональной терапии.
МКБ-10
Общие сведения
Синдром андрогенодефицита у мужчин встречается в разные возрастные периоды жизни: от пубертата до зрелости. Возрастной андрогенный дефицит обусловлен недостаточным количеством андрогенов в организме мужчины. Это состояние является естественным возрастным изменением, и первые его симптомы начинают проявляться после 50-ти лет, ранний андрогенный дефицит и симптомы преждевременного старения проявляются в 40-45 лет. Андрогенная недостаточность возникает из-за уменьшения продукции тестостерона, что сказывается на всех органах и системах.
Причины
Тестостерон является основным мужским гормоном. Он отвечает за формирование мужского типа поведения и вторичных половых признаков (рост волос на лице, огрубение голоса). Рост и развитие половых органов, прибавка мышечной массы, эрекция невозможны без тестостерона. Жировая клетчатка у мужчин распределяется совершенно не так, как у женщин; сперматогенез и развитие скелета по мужскому типу тоже полностью зависят от уровня тестостерона в организме.
Гипофиз и гипоталамус вырабатывают гормоны, благодаря которым тестостерон продуцируется в яичках клетками Лейдинга. Оба этих процесса являются взаимозависимыми и регулируются в зависимости от необходимости и от процентной концентрации каждого гормона. Весь тестостерон находится в крови в активных и неактивных фракциях; за основные действия тестостерона отвечает гормон, находящийся в активных фракциях.
Точная причина андрогенного старения неизвестна, но из всех теорий, которые имеет на сегодняшний день клиническая андрология, несколько являются наиболее вероятными. Возрастные атеросклеротические изменения в организме ведут к снижению кровоснабжения в области тестикул, что влияет на размер и состояние клеток Лейдинга – они становятся менее активными. По этой же теории в определенное время наступает апоптоз клеток Лейдинга, то есть их запрограммированная гибель.
В гипоталамусе и гипофизе с возрастом так же происходят дистрофические и склеротические изменения, что приводит к нарушениям регуляции процесса выработки тестостерона. Теория о том, что наследственная предрасположенность может играть основную роль в развитии раннего андрогенного дефицита, подтверждается тем, что активность гормонпродуцирующей ткани является генетически обусловленной.
Симптомы андрогенного дефицита
Физиологическое старение из-за снижения уровня тестостерона в крови проявляется в первую очередь со стороны репродуктивной системы: снижается половое влечение (либидо), возможна слабая эрекция и отсутствие эякуляции после полового акта, у некоторых мужчин оргазм становится менее ярким. Все это ведет к физиологическому возрастному бесплодию, если андрогенный дефицит не является преждевременным.
Со стороны других органов и систем степень выраженности симптомов зависит от степени гипогонадизма. У большинства мужчин появляются вегето-сосудистые расстройства: не связанные с работой надпочечников колебания артериального давления, приливы, чувство нехватки воздуха, головокружения и беспричинные покраснения кожи шеи и верхней половины груди.
Нарастают и психоэмоциональные нарушения, что проявляется быстрой утомляемостью, нарушениями сна, раздражительностью и депрессиями. У мужчин уменьшается объем мышечной массы, иногда происходит ее замещение на жировую, при этом жировая ткань откладывается повсеместно, а не только в подкожном слое. У некоторых мужчин незначительно увеличиваются в размерах молочные железы, и уменьшается рост волос в области лица. Более серьезным проявлением гипогонадизма является снижение плотности костной ткани (остеопороз), что может стать причиной частых переломов.
Осложнения
Снижение тестостерона в крови повышает вероятность онкологических заболеваний предстательной железы, вероятность возникновения сахарного диабета, а также ведет к прогрессированию атеросклероза сосудов с высоким риском инсульта сосудов головного мозга и инфаркта миокарда.
Диагностика
Жалобы пациента, особенности во внешности и определение концентрации тестостерона являются основными диагностическими критериями. Анкеты, с помощью которых гораздо проще провести оценку жалоб пациента, кроме того и более точны, так как пациент может не говорить вслух о своих проблемах. Опросник AMS определяет выраженность симптоматики в баллах, но дополнительно необходимо пройти лабораторное обследование, чтобы выявить уровень андрогенного дефицита.
В лаборатории исследуют сыворотку крови на содержание общего тестостерона и биологически активного тестостерона. Проведут общий и биохимический анализ крови, а так же определят количество глобулина, связывающего половые стероиды (ГСПС).
Нормальный показатель общего тестостерона в сыворотке крови не менее 12 нмоль/л, при этом сразу после получения результатов можно говорить о соответствии предварительного диагноза и действительности. Но в некоторых случаях уровень общего тестостерона в пределах нормы, а содержание свободного гормона снижено из-за избыточного его связывания с белками крови. Тогда прибегают к исследованию количества свободного тестостерона в сыворотке крови.
Всем мужчинам, у которых есть вероятность андрогенного дефицита также необходимо пройти обследование костной ткани на плотность (денситометрия) и другие исследования, точный перечень которых определит лечащий врач-андролог.
Лечение андрогенного дефицита
Основным методом коррекции андрогенного дефицита является заместительная терапия с использованием различных препаратов тестостерона. Иногда прибегают к стимуляции выработки собственного тестостерона с помощью хорионического гонадотропина.
Заместительная терапия проводится только после исключения онкологических заболеваний предстательной железы. Поэтому важно проходить раннюю лабораторную диагностику по достижению 40-45 лет. Так как недостаток тестостерона повышает вероятность возникновения карциномы предстательной железы, а начатая заместительная терапия лишь ухудшает состояние пациента, хотя при нормальном уровне тестостерона в сыворотке крови вероятность развития карциномы предстательной железы в несколько раз ниже. Чтобы исключить онкозаболевания предстательной железы, в лаборатории определяют уровень простатоспецифического антигена, и при необходимости проводят более детальное обследование на наличие раковых клеток.
Заместительные препараты необходимо принимать пожизненно, так как яичники уже не вырабатывают тестостерон, пациентам необходимо периодически проводить контроль состояния предстательной железы и определять уровень ПСА.
Эффективность лечения становится заметной после накопления в организме нужной концентрации тестостерона и устранения проявлений его недостатков. При этом заместительная терапия полностью безопасна, имеет минимум побочных эффектов и позволяет мужчинам особенно с преждевременным андрогенным дефицитом вести привычный образ жизни и сохранить активность.
Способов введения тестостерона в организм человека несколько. Существуют пластыри, кремы или гели – в этом случае тестостерон поступает трансдермально, подкожные импланты являются препаратами пролонгированного действия, что очень удобно, так как нет необходимости следить за ежедневным принятием тестостерона. Классическими формами являются таблетированные формы для перорального приема и масляные растворы для внутримышечных инъекций.
1. Возрастной андрогенный дефицит: клиника, диагностика, лечение и мониторинг/М.А. Франк, И.В. Борзунов, А.И. Гомжин, М.О. Мурзин. - 2016.
2. Рекомендации по диагностике и лечению дефицита тестостерона (гипогонадизма) у мужчин/ И.И. Дедов, Г.А. Мельниченко, Р.В. Роживанов, Д.Г. Курбатов. - 2016.
Синдром Морриса - симптомы и лечение
Что такое синдром Морриса? Причины возникновения, диагностику и методы лечения разберем в статье доктора Литвинова Владимира Валентиновича, репродуктолога со стажем в 39 лет.
Над статьей доктора Литвинова Владимира Валентиновича работали литературный редактор Юлия Липовская , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Морриса (синдром тестикулярной феминизации) — это врождённое генетическое заболевание, при котором у людей мужского пола ткани-мишени не чувствительны к мужским половым гормонам — андрогенам. Человек с синдромом Морриса генетически является мужчиной (имеет кариотип 46 XY), но выглядит как женщина.
Кариотип — это набор хромосом, который передаётся ребёнку от матери и отца. Кариотип позволяет определить характеристики индивида, включая пол. В норме у человека в генетическом наборе присутствует 46 хромосом, из них 22 пары аутосомных (не определяющих пол) и одна пара половых хромосом, которая определяет гендерную принадлежность ребёнка. Половые хромосомы женщины обозначаются как ХХ, мужчины — ХY. То есть нормальный женский кариотип — 46 XX, мужской — 46 XY.

Причина синдрома Морриса — мутации (изменение) гена рецептора андрогенов, у наследованные от матери или возникшие впервые . Мутации обуславливают резистентность (нечувствительность) рецепторов к гормону тестостерону [19] . В этом случае развивается синдром тестикулярной феминизации (СТФ). Ещё это заболевание называют синдромом Морриса — по имени американского гинеколога, который впервые ввёл термин в обиход, подробно описав его в 1953 году. Другие синонимы патологии — синдром нечувствительности к андрогенам, синдром андрогенной резистентности.
Часто мужчины с синдромом Морриса даже не догадываются о своём биологическом поле (как и их родители) и живут как девочки/женщины. Это объясняется тем, что данная патология больше никак себя не проявляет, кроме проблем с фертильностью (способностью к зачатию) во взрослом возрасте.
В популяции синдром встречается редко. По данным разных авторов, частота заболевания составляет от 1: 20 400 до 1: 99 100 случаев [19] . Точные данные собрать сложно, так как патология часто остаётся нераспознанной. Согласно исследованию, которое проводилось в Дании в течение семилетнего периода, полная нечувствительность к андрогенам случается с вероятностью 1 на 20 400 новорождённых с кариотипом 46 XY [15] .
Синдром тестикулярной феминизации впервые в Европе описан в 1817 году баварским врачом Джорджем Стегленером, в России — в 1893 году профессором клиники московского университета Благоволиным Сергеем Ивановичем (1865-1947) [4] .

Есть заключения генетиков и историков (по утверждению профессора Эфроимсона), которые приписывают синдром тестикулярной феминизации и королеве Англии Елизавете Тюдор (1533-1603) [5] .
Несмотря на то, что синдром Морриса встречается редко, он обнаруживается почти у 1 % выдающихся спортсменок , которые имеют превосходство в физической силе, быстроте и ловкости [5] [18] . Это стало причиной исключения женщин и девушек с синдромом тестикулярной феминизации из женских спортивных состязаний [5] . Регламент многих серьёзных спортивных соревнований, особенно в силовых видах спорта, беге, прыжках, обязывает женщин предоставить результат анализа на кариотип. Обязательное тестирование стали проводить после истории с немецкой легкоатлеткой Дорой Ратьен, которая участвовала в Олимпийских играх 1936 года. После игр стало известно, что Дора генетически была мужчиной. Этот случай послужил поводом к тому, что всех участниц соревнований стали осматривать врачи. Позже вместо осмотров спортсменки стали сдавать анализ крови на кариотип.

Иногда синдром Морриса называют синдромом манекенщиц , так как женщины с данной патологией часто имеют привлекательную внешность [16] [17] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Морриса
Как правило, люди с синдромом тестикулярной феминизации имеют женский фенотип, т. е. внешне выглядят как женщины. Однако есть некоторые признаки, по которым можно определить наличие заболевания.
- С началом полового созревания больные отличаются женским телосложением. Это объясняется влиянием эстрогенов и отсутствием эффекта тестостерона. П ри этом у таких людей часто высокий рост, узкий таз и широкие плечи, что более характерно для мужчин. [1][2][6][7] .

- При нормальном росте и отсутствии аномалий развития характерны крупные кисти рук и крупные стопы.
- Молочные железы обычно развиты соответственно 3-4 размеру, однако ареолы сосков окрашены бледно.
- Характерно отсутствие полового оволосения (в области подмышечных впадин и на лобке), т. к. "не работает" тестостерон.
- В результате влияния эстрадиола и отсутствия действия тестостерона в ходе эмбрионального развития у плода мужского пола наружные половые органы формируются по женскому типу: не формируется половой член, но есть половые губы, укороченное влагалище, заканчивающееся слепо (т. е. нет матки, шейки матки и яичников).
- У детей с синдромом Морриса (4-5 лет) иногда появляются паховые грыжи. В таких случаях родители обращаются к детскому хирургу. Врач проводит операцию и в составе грыжевого содержимого обнаруживает ткань, напоминающую по структуре яичко (мужскую гонаду). Так как у "девочки" не может быть мужской гонады, берут биопсию (для подтверждения диагноза СТФ). В таком случае яичко не удаляют, ушивают грыжевой мешок и заканчивают операцию. Далее врач рекомендует провести генетическое обследование ребёнка, которое устанавливает мужской кариотип 46 XY. Считается, что удалять гонады до пубертатного периода нельзя, т. к. это может нарушить формирование организма ребёнка в целом.
- В 14-16 лет (если патология не была выявлена раньше), родители замечают, что у "девочки" отсутствует менструация, в связи с чем обращаются к врачу-гинекологу. Во время гинекологического осмотра и УЗИ выявляется, что у ребёнка имеется слепо заканчивающееся влагалище (в виде слепого мешка). Глубина влагалища может варьировать от нормальной до укороченной [1][2][6][7] .
Патогенез синдрома Морриса
В начале эмбрионального развития у зародышей вне зависимости от хромосомного набора, образовавшегося при оплодотворении яйцеклетки сперматозоидом, половая система закладывается одинаково и предоставляет возможности для развития как женской, так и мужской половой системы. В частности, у зародыша одновременно формируются вольфов и мюллеров протоки, которые потом превращаются в семявыносящие протоки у мужчин и матку с фаллопиевыми трубами и влагалищем у женщин. Половые железы эмбриона (гонады) не дифференцированы и содержат первичные половые клетки (гаметы), которые могут превратиться как в клетки яичников (женские гонады), так и в клетки семенников (мужские гонады).
Таким образом, эмбрион до 6 недель является нейтральным по полу (имеет признаки и мужского, и женского пола). Далее процесс формирования половых признаков и в дальнейшем организма происходит под строгим контролем гормонов: у эмбриона мужского пола — под влиянием тестостерона (влагалище атрофируется), у эмбриона женского пола — под влиянием эстрадиола и прогестерона (влагалище трансформируется из "слепого мешка" и формируется шейка матки и матка). Тестостерон и эстрадиол вырабатываются как у мужчин, так и у женщин, только их соотношение разное. В норме у мальчиков соотношение мужского гормона к женскому 4\1, а в случае СТФ — 0\1. У девочек соотношение мужского гормона к женскому, прямо противоположное — 1\4.

В процессе эмбриогенеза у людей с синдромом Морриса под влиянием мужской Y-хромосомы гонады развиваются как яички, они не способны к сперматогенезу, но способны вырабатывать тестостерон [8] . Однако из-за генетической мутации организм "не видит"/"не чувствует" присутствие тестостерона в крови, поэтому гормон не может проявить свои свойства и сформировать мужской организм. При этом секретируемые надпочечниками эстрогены (эстрадиол), хоть и в небольшом количестве, нормально вырабатываются и усваиваются организмом, в результате чего начинают превалировать в организме (как у девочки). В связи с этим влагалище не атрофируется, в дальнейшем ребёнок развивается по женскому типу.
Классификация и стадии развития синдрома Морриса
Синдром тестикулярной феминизации делят на две формы — полный и неполный. Дети с полной формой нечувствительности к мужским гормонам имеют однозначно женский внешний вид. При этом состоянии чувствительность организма к мужскому половому гормону (тестостерону) отсутствует полностью. Рождается здоровая "девочка", не имеющая, на первый взгляд, каких-либо отклонений в развитии.
Неполная форма СТФ характеризуется более разнообразной клинической картиной. В этом случае отмечается некоторая чувствительность рецепторов к тестостерону. Выделяют пять основных степеней неполного синдрома тестикулярной феминизации (классификация 1996 года) [10] .
- Мужской тип. У больных мужской фенотип без каких-либо отклонений.
- Преимущественно мужской тип. Наблюдаются нарушения формирования половых органов, хотя фенотип больных — мужской.
- Амбивалентный тип. Более выраженные нарушения формирования половых органов: уменьшение полового члена, который становится похожим на клитор, мошонка похожа на большие половые губы. Характерно расширение таза, узкие плечи, гинекомастия (увеличение груди).
- Преимущественно женский тип. По фенотипическим признакам больные являются женщинами. Однако у них, как правило, короткое влагалище и гипертрофированый (увеличенный) клитор
- Женский тип. По всем внешним признакам, за исключением увеличенного клитора, больные являются женщинами.

Осложнения синдрома Морриса
- Из-за нарушения проходимости (опущения) яичек по паховому каналу у больных СТФ в детском возрасте (3-5 лет) часто обнаруживают паховые грыжи и гипоспадию ( недоразвитие полового члена и неправильное расположение мочеиспускательного канала) . Гипоспадия, в свою очередь, может стать причиной развития различных воспалительных процессов в мочевыделительной системе ( уретриты , пиелонефриты) [13] .

- Крипторхизм (неопущение яичек) в будущем грозит злокачественным перерождением тканей яичка (развитием гонадобластомы), что является наиболее тяжёлым осложнением данного заболевания [10][19] . Одна из причин образования гонадобластомы в том, что яички постоянно находятся в забрюшинном пространстве, где температура выше 37 °C. В норме яички должны находиться снаружи (в мошонке), т. к. для их жизнедеятельности нужна постоянная температура ниже 34 °C. Точную частоту возникновения рака у пациентов с синдромом тестикулярной феминизации оценить очень трудно, однако по различным данным общий риск составляет примерно 5 % всех случаев патологии. При этом с возрастом вероятность развития рака возрастает [reference:19 ] .

- Ещё одним серьёзным осложнением заболевания является бесплодие.
Диагностика синдрома Морриса
- Определение фенотипа — совокупности внешних признаков.
- Изучение семейного анамнеза пациента. Если в семье были случаи андрогенной нечувствительности, то вероятность наличия патологии выше, однако нужно понимать, что отсутствие отягощённого семейного анамнеза не исключает диагноз.
- Гинекологический и урологический осмотр. Выявляется слепо заканчивающееся влагалище (слепой мешок), отсутствие шейки, не пальпируется матка и её придатки.
- Определение половых гормонов в крови (тестостерона, эстрадиола). Выявляется высокий уровень тестостерона, уровень эстрадиола выше, чем у мужчин, но ниже, чем у женщин в норме.
- Ультрасонографии органов малого таза (УЗИ) и р ентгенологическое обследование для выяснения состояния органов малого таза . В малом тазу не визуализируется матка и яичники. Забрюшинно визуализируются образования, похожие на "яичники", на самом деле это яички (без дополнительных данных анамнеза бывает трудно это предположить). Мужские гонады могут располагаться в паховых каналах, в стенках таза или в толще больших половых губ.
- Кариотипирование — исследование хромосомного набора. Позволяет обнаружить отклонения в структуре и числе хромосом . При синдроме тестикулярной феминизации определятся мужской кариотип — 46 XY. Для исследования используется венозная кровь.
- Молекулярно-генетический анализ гена андрогенного рецептора. Определение мутаций гена при наличии характерной клинической картины подтверждает диагноз СТФ с вероятностью близкой к 100 %.
Лечение синдрома Морриса
Лечение синдрома тестикулярной феминизации должно осуществляться междисциплинарной командой врачей, которая состоит из хирурга, гинеколога, генетика, эндокринолога и клинического психолога или психиатра.
При полной андрогенной нечувствительности и в случае частичной нечувствительности с преимущественно женским типом в 20-50 % случаев необходимо удалять яички из-за риска развития рака [19] . Операция, как правило, проводится после завершения пубертатного периода и конституционального формирования (14-15 лет) [11] . Хирургическое лечение в отношении половых желёз (гонад) проводится в настоящее время лапароскопическим доступом [3] [6] . Лапароскопию должен проводить хирург высокой квалификации, так как удаляемые гонады (яички) находятся забрюшинно и имеют высокую степень кровоснабжения. Динамическое наблюдение, УЗИ и контроль гормона тестостерона позволяют оценивать эффективность проведённой операции [14] .
Операция эффективна в 100 % случаев. Но так как удаляемые гонады мягкие и не имеют чёткой формы и структуры, возможны случаи неполного их удаления. Тогда оставшаяся ткань яичка может возобновить свою работу, что проявляется повышением тестостерона в крови и визуализацией на УЗИ гонады/гонад. В этом случае необходима повторная операция. Однако это случается очень редко.
После удаления гонад показана последующая длительная заместительная терапия эстрогенами. Она необходима для продолжения дальнейшего формирования женского организма и обязательна (до возраста 45-50 лет) для профилактики остеопороза (потери костной тканью кальция) [8] [12] . Развитие остеопороза связано с тем, что в организме пациента мало собственного эстрадиола, который в норме удерживает кальций в костях. Следовательно, у таких людей кальций будет вымываться быстрее и плотность костей будет снижаться. Такое состояние опасно частыми переломами.
В случае короткой длины влагалища для предотвращения диспареунии (боли во время полового акта) возможно хирургическое увеличения длины влагалища [20] .
Больным с частичной андрогенной нечувствительностью с преимущественно мужским типом , которые выросли как мужчины, может быть предложено хирургическое лечение крипторхизма и гипоспадии. При крипторхизме проводится орхипексия — низведении неопущенного яичка в мошонку и фиксации его к окружающим тканям путём наложения шва. Коррекция гипоспадии подразумевает:
- восстановление отсутствующей части мочеиспускательного канала;
- восстановление нормального расположения мочеиспускательного канала;
- выпрямление полового члена;
- придание эстетически адекватного внешнего вида наружным половым органам.

В случае гинекомастии (увеличения груди) мужчинам может потребоваться маммопластика [21] . Часто пациентам с выявленным заболеванием требуется психологическая помощь.
Прогноз. Профилактика
Прогноз для жизни в случае синдрома Морриса благоприятный. Своевременная диагностика, гонадэктомия и заместительная гормональная терапия обычно дают хорошие краткосрочные и долгосрочные результаты. Пациентки часто адаптируются, нередко выходят замуж. Преодоление бесплодия возможно с помощью программ ЭКО с использованием донорских яйцеклеток, оплодотворённых спермой мужа, и суррогатного материнства. Главное — правильная адаптация и объяснение пациентке её состояния [12] .
Для профилактики синдрома тестикулярной феминизации женщинам, планирующим беременность рекомендуется пройти генетическое исследование, чтобы выяснить, не является ли она носителем патологического гена. Исследование должно проводится при отягощённом наследственном анамнезе, т. е. в том случае, если в семье были случаи андрогенной нечувствительности. Для профилактики остеопороза рекомендуется дополнительный приём кальция и витамина D [8] .
Синдром нечувствительности к андрогенам. Причины и диагностика
Аменорея при синдроме нечувствительности к андрогенам. Диагностика и лечение
Синдром полной нечувствительности к андрогенам — третья по частоте причина первичной аменореи после дисгенезии гонад и агенезии мюллеровых протоков. Он встречается с частотой от 1:40 000 до 1:99 000 и был впервые описан Моррисом в 1953 г.. Больные подростки имеют кариотип 46.XY. В 85-90% случаев синдрома выявляют мутацию гена андрогеновых рецепторов (АР).
Мутация локализуется на Х-хромосоме, Xqll-12, и способна вызывать широкий спектр нарушений вирилизации, от легкой нечувствительности к андрогенам до полной формы. К настоящему времени идентифицировано более 300 мутаций гена АР Х-хромосомы.
У таких больных наружные половые органы развиваются по женскому типу из-за неспособности распознавать андрогены. Однако, благодаря наличию яичек и активному синтезу антимюллерового гормона (АМГ), производные мюллеровых протоков обычно редуцируются. У больных имеются нормально развитые женские наружные гениталии, заканчивающиеся слепым влагалищным карманом. Хотя большинству больных диагноз ставят в период полового созревания и лишь иногда — при рождении или в детстве, подозрение на этот синдром появляется, когда у девочек без признаков развития гениталий по промежуточному типу обнаруживают одно- или двусторонние паховые грыжи.
Рост больных в пределах нормы или несколько выше. Развитие молочных желез у них завершено, но при ближайшем рассмотрении становится заметно, что выглядят они необычно. Соски, как правило, маленькие, ареолы бледные. Волосы на лобке и в подмышечных областях редкие или отсутствуют. Возможно недоразвитие малых половых губ. Лабораторно выявляют нормальное для лиц мужского пола количество тестостерона и несколько повышенное содержание ЛГ. При визуализации органов малого таза обнаруживают отсутствие матки.
Важно определить расположение яичек. Они могут находиться интраабдоминально или в паховом канале. Несмотря на наличие канальцев в яичках, признаки сперматогенеза в них отсутствуют. Из-за высокого риска развития новообразований половых желез таким больным показана гонадэктомия. Когда проводить гонадэктомию, четких рекомендаций нет. Некоторые специалисты рекомендуют удалять половые железы после развития молочных желез до Ма.3-4 по Таннеру, если диагноз был установлен до пубертата или в периоде полового созревания.
Подростки легче переносят изменения, связанные с половым созреванием, на собственном гормональном фоне, нежели чем на фоне гормональной терапии. Возможность развития опухоли в период ожидания невелика. Другие врачи выступают за удаление половых желез сразу же после подтверждения диагноза с целью профилактики потенциальной андрогенизации, возможной в процессе полового созревания. Если гонадэктомия проведена в пубертате, пациентку и членов ее семьи следует проинформировать о необходимости эстрогенотерапии для поддержания развития молочных желез и сохранения нормального метаболизма костной ткани.

Дифференциальная диагностика синдрома нечувствительности к андрогенам
Синдром нечувствительности к андрогенам следует дифференцировать от синдрома Майера— Рокитанского—Кюстера—Хаузера. И в том, и в другом случае у пациенток имеется первичная аменорея и нормальное развитие молочных желез. Однако во втором случае обнаруживают кариотип 46,ХХ и яичники. Очень информативным показателем служит концентрация тестостерона: у пациенток с синдромом нечувствительности к андрогенам она находится в диапазоне, характерном для мужчин. После выявления повышенного содержания тестостерона для подтверждения диагноза проводят кариотипирование.
Лечение синдрома нечувствительности к андрогенам
Обычно у пациенток присутствует слепой вагинальный карман. Таким образом, может понадобиться удлинение влагалища путем последовательной дилатации кармана. Хирургическое вмешательство обычно не показано. В одном из долгосрочных исследований, проведенных в Университете им. Джона Хопкинса в США, наблюдали за 14 женщинами, страдающими синдромом полной нечувствительности к андрогенам, в течение десятилетий. Возраст женщин находился в диапазоне от 25 до 65 лет. Более чем половина из них не нуждались в удлинении влагалища.
Все женщины были удовлетворены своим полом, 71% сообщили об удовлетворении своей сексуальной функцией, 77% испытывали оргазм.
Более половины женщин с трудом представляли себе, что такое синдром нечувствительности к андрогенам. Но только половина из этих женщин высказывали по этому поводу сожаление. Данное исследование подтверждает важность длительного наблюдения и поддержки подростков по мере их перехода во взрослый контингент.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Существует две формы синдрома: частичная и полная. Какая из них развивается, зависит от остаточной функции андрогеновых рецепторов (АР). Синдром полной нечувствительности к андрогенам характеризуется остановкой развития производных мюллеровых протоков. При этом нет промежуточного типа развития гениталий, хотя налицо признаки тестикулярной феминизации: нормально развитые половые губы, клитор и вход во влагалище. Иногда можно пропальпировать яички в паховых областях.
Если такое случается у ребенка с женским фенотипом, важно уточнить, что же именно пальпируется: 7% новорожденных девочек с паховой грыжей и отсутствием шейки матки при пальпации или вагиноскопии страдают мужским псевдогермафродитизмом, как правило, на фоне синдрома нечувствительности к андрогенам, или страдают смешанной дисгенезией гонад. При синдроме частичной нечувствительности к андрогенам обнаруживают гениталии промежуточного типа с различной степенью развития вольфовых структур и губно-мошоночного сращения.
Патогенез синдрома нечувствительного к андрогенам
Дефект действия андрогенов развивается вследствие мутации гена Андрогеновых рецепторов (АР), нарушающей связывание тестостерона и ДГТ в андроген-чувствительных органах-мишенях. Ген АР подвергается нарушающей его функционирование мутации в локусе Xqll-13. Пострецепторные процессы, осуществляющие эффект андрогенов в тканях-мишенях, не происходят, несмотря на нормальный синтез андрогенов, из-за чего пренатально развивается недостаточная вирилизация наружных гениталий плода.
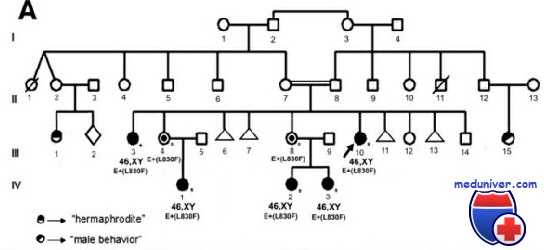
Описано более 250 мутаций, в том числе полные и частичные делеции, точечные мутации и микроинсерции/микроделеции. Как и предполагалось, набор функциональных дефектов может быть обусловлен любой из них. Некоторые приводят к полной утрате рецепторов клеточных мембран, в то время как другие — к неполноценному синтезу белка вследствие изменений аффинности связывания субстрата. Изменения аффинности могут вносить вклад в ухудшение передачи сигнала, несмотря на наличие нормальных рецепторов клеточных мембран.
При синдроме полной нечувствительности к андрогенам генотип соответствует фенотипу в большинстве случаев. Это, однако, не так при синдроме частичной нечувствительности к андрогенам. Нормальное количество антимюллерова гормона (АМГ) обусловливает редукцию мюллеровых протоков. Однако структуры — производные вольфовых протоков развиваются неправильно вследствие уменьшения или отсутствия связывания тестостерона и ДГТ.
Диагностика синдрома нечувствительного к андрогенам
Концентрация гормонов соответствуют возрастной норме для мужчин. Однако при пробе со стимуляцией ХГЧ повышается содержание как тестостерона, так и ДГТ. Подтверждение диагноза проводят с помощью анализа мутации гена АР, при котором определяют более 95% мутаций, связанных с синдромом полной или частичной нечувствительности к андрогенам, или с помощью определения связывания субстрата с АР клеток кожи мошонки, которое должно быть снижено при этом заболевании.
Для идентификации мюллеровых структур проводят УЗИ. Если эти структуры обнаруживают, диагноз синдрома полной или частичной нечувствительности к андрогенам исключают.
Дефекты синтеза андрогенов. Причины и диагностика
Нарушения на любом из этапов ферментативного биосинтеза тестостерона из холестерина могут приводить к изменениям андрогенного профиля. У развивающегося плода мужского пола это может привести к образованию гениталий промежуточного типа. Как и большинство энзимных дефектов, наследуются эти заболевания по аутосомно-рецессивному типу.
Врожденная липоидная гиперплазия надпочечников редкая тяжелая форма врожденной гиперплазии коры надпочечников (ВГКН), заканчивающаяся летально у двух третей пораженных новорожденных детей. Сущность ее заключается в абсолютном дефиците всех стероидных гормонов надпочечников. У новорожденных имеются наружные гениталии женского типа и выраженная потеря соли. При рождении обнаруживают симптомы тяжелой надпочечниковой недостаточности, включающие синдром задержки роста, рвоту, диарею, гипонатриемию и гипокалиемию.
Патогенез и диагностика врожденной липоидной гиперплазии надпочечников. Врожденная липоидная гиперплазия надпочечников может быть обусловлена мутациями двух разных генов. Мутации гена, кодирующего стероидогенныи активный регуляторный протеин, бывают причиной развития этого синдрома в большинстве случаев. Этот белок служит посредником быстрого входа холестерина в митохондрии, являясь экстренным регулятором стероидогенеза.
Реже причиной развития синдрома выступают мутации гена CPY11A, кодирующего фермент расщепления боковой цепи холестерина 20,22-десмолазу. При предварительных лабораторных исследованиях у детей выявляют снижение содержания глюкокортикоидов, минералокортикоидов, тестостерона и ДГТ наряду с уменьшением содержания натрия и калия. Большинство детей погибают до окончания периода новорожденности. Выжившие должны постоянно получать заместительную терапию глюко- и минералокортикоидами.
Недостаточность 3b-гидроксистероиддегидрогеназы как причина нарушения синтеза андрогенов. Одна из форм врожденной гиперплазии коры надпочечников, которая может проявляться как женским, так и мужским псевдогермафродитизмом. Подробно описана в разделе этой главы, посвященном женскому псевдогермафродитизму.
Недостаточность 17a-гидроксилазы как причина нарушения синтеза андрогенов
Недостаточность 17а-гидроксилазы характеризуется снижением количества, вплоть до полного отсутствия, половых гормонов, синтезируемых как гонадами, так и надпочечниками, при одновременном повышении синтеза минералокортикоидных предшественников. У новорожденных мужского пола половые органы развиты по промежуточному типу, в то время как у новорожденных девочек отмечают недоразвитие половых органов.
Заболевание сопровождается артериальной гипертензией различной степени выраженности и гипокалиемией. У девочек недостаточность 17а-гидроксилазы зачастую диагностируют в раннем подростковом возрасте при обследовании по поводу задержки полового развития, отсутствия вторичных половых признаков или первичной аменореи, хотя в некоторых случаях диагностика возможна уже при рождении.

Мутации гена CYP17 могут клинически проявляться в виде недостаточности 17а-гидроксилазы, 17,20-лиазы или их сочетания. При недостаточности 17а-гидроксилазы снижение содержания кортизола стимулирует синтез кортикотропина, и, хотя продукция стероидов повышается, она все равно блокируется на этапе 17а-гидроксилазы. Компенсаторно накапливаются 17-дезоксистероиды, в том числе прегненолон, прогестерон, дезоксикортикостерон и кортикостерон.
Снижение синтеза андрогенов приводит к гипогонадизму. Вследствие минералокортикоидной активности дезоксикортикостерона возникают гипернатриемия и потеря калия, увеличивается объем плазмы крови, развивается артериальная гипертензия. Обычно выявляют гипокалиемию, на фоне которой снижаются количество альдостерона сыворотки крови и активность ренина плазмы.
Диагноз подтверждают при выявлении выраженного повышения содержания 11-дезоксикортикостерона и кортикостерона сыворотки. Количество прегненолона и прогестерона слегка повышено.
Сывороточные концентрации следующих стероидов ничтожны или совсем не определяются: 17а-гидроксипрегненолона, 17а-гидроксипрогестерона, 11-дезоксикортизола, ДГЭА-сульфата, андростендионаитестостерона. Концентрации эстрогенов в сыворотки крови и моче снижены, а кортикотропина, ФСГ и ЛГ — повышены. Выводимые с мочой метаболиты (17а-гидроксикортикостероиды и 17-кетостероиды) имеют недостаточную концентрацию или отсутствуют.
Возможна пренатальная диагностика: определение содержания стероидов надпочечников в амниотической жидкости.
Недостаточность 17,20-десмолазы (лиазы) как причина нарушения синтеза андрогенов
При этом синдроме блокируются пути превращения 21-углеродных стероидов (17-гидроксипрегненолон и 17-гидроксипрогестерон) в 19-углеродные стероиды, ДГЭА-С и андростендион соответственно. Отмечают также снижение количества андрогенов, тестостерона и эстрадиола. Блокада может быть как полной, так и частичной, в зависимости от этого могут различаться клинические симптомы.
Как уже было сказано выше, ген CYP17 кодирует 17а-гидроксилазно-17,20-лиазный комплекс, а его дефекты могут приводить к недостаточности 17,20-десмолазы (лиазы). Лабораторно выявляют повышение концентрации 17-гидроксипрегненолона сыворотки, 17-гидроксипрогестерона, нормальное содержание ФСГ при повышении ЛГ, снижение тестостерона и эстрадиола, ДГЭА-сульфата, андростендиона, а также нормальное или слегка повышенное содержание прегненолона и прогестерона.

Недостаточность 17b-гидроксистероиддегидрогеназы как причина нарушения синтеза андрогенов
Диагноз этого заболевания зачастую ставят в период полового созревания лицам с мужским генотипом. Они могут воспитываться как девочки и обращаться с жалобами на отсутствие менструаций и гирсутизм, или как мальчики, и тогда обращаться с жалобами на гинекомастию и недоразвитие половых органов. У пораженных лиц мужского пола показатели вирилизации, включая клиторомегалию вплоть до микрофалоса и развитие вторичных мужских половых признаков в период полового созревания, очень напоминают таковые при недостаточности 5а-редуктазы. Все больные бесплодны.
Фермент 17b-гидроксистероиддегидрогеназа, или 17-кетостероидредуктаза, катализирует превращение андростендиона в тестостерон в яичках. Мутации гена изофермента 17b-гидроксистероиддегидрогеназы типа 3 (HSD17B3), расположенного на хромосоме 9q22, приводят к недостаточному синтезу тестостерона. Большинство их представлено миссенс/нонсенс-мутациями. Существенной корреляции между генотипом и фенотипом не выявляют.
Развитие гениталий по промежуточному типу обычно выявляют при рождении. Чаще всего имеются кли-торомегалия, сращение губно-мошоночных складок и слепой влагалищный карман. Яички нередко пальпируются в паховых каналах или губно-мошоночных складках, хотя иногда могут быть расположены и в брюшной полости. Как и при других формах мужского псевдогермафродитизма, внутренние отделы мочеполового тракта развиты нормально.
Есть придатки яичка, семявыносящие протоки, семенные пузырьки, семявыбрасывающие протоки. Предстательная железа и производные мюллеровых протоков отсутствуют.
Характерной лабораторной находкой бывает повышенное соотношение андростендион/тестостерон, образующееся в результате увеличения количества андростендиона и снижения тестостерона. Возможна пренатальная диагностика у потомства пораженных пациентов, если у последних выявлена специфическая мутация.
Читайте также:
- Желудочно-кишечный тракт пожилых. Особенности
- Нарушения привычек и влечений (импульсного контроля)
- Лучевая диагностика эпидермоидной кисты позвоночника и спинного мозга
- Вегетативные расстройства при контузиях. Изменения крови при контузиях
- Диагностика гардренелл. Микробиологическая диагностика гардренеллеза. Лечение гардренеллеза.