Механизм активации онкогенов и протоонкогенов клетки
Обновлено: 03.05.2024
Протоонкогены превращаются в онкогены под влиянием канцерогенных агентов - физических, химических, биологических (онковирусов). Возможно участие нескольких основных механизмов активации протоонкогена: включение промотора, амплификация, транслокация, инсерции, трансдукция, мутация.
Включение промотора. Промотором считается участок ДНК, с которым связывается РНК-полимераза, когда запускается синтез гена (или онкогена). Условием, необходимым для проявления активирующего действия промотора, является расположение его вблизи протоонкогена, что обеспечивало бы его взаимодействие с выше указанным протоонкогеном. Роль промоторов для протоонкогенов могли бы играть ДНК-копии определенных участков генома онкорнавирусов. Подтверждением сказанному могут служить опыты с индуцированием определенных опухолей, например, лимфомы у птиц введением ретровируса ALV, лишенного онкогена. В этом случае ДНК-копия РНК ретровируса включалась около протоонкогена c-myc, что сопровождалось увеличением транскрипции им РНК в 20-100 раз.
Роль промоторов для протоонкогенов могут исполнять мобильные генетические структуры (так называемые «прыгающие» гены), представляющие собой фрагменты или участки ДНК, способные перемещаться и встраиваться в разные участки генома клетки. Перемещающиеся генетические фрагменты - мобильные гены, обнаружены в клетках практически всех животных, включая человека. Сейчас стало известно, что мобильные гены и протовирусы (ДНК-копии онкорнавирусов) - это элементы генома, сходные между собой.
Амплификация. Под этим термином понимают приумножение числа, или образования дополнительного количества копий протоонкогенов, обладающих в норме крайне низкой, следовой активностью. Как результат такого многократного увеличения протоонкогенов общая активность последних значительно возрастает, что может привести к опухолевой трансформации. В качестве примера можно привести наблюдения над развитием промиелоцитарного лейкоза, когда дополнительное увеличение числа копий протоонкогена c-myc до 16-32 приводило к опухолевой трансформации. Еще одним подтверждением роли амплификации протоонкогенов в опухолевой трансформации клеток могут служит опыты, в которых показана возможность индукции перехода нормальной клетки в опухолевую при введении в клеточный геном большого числа (до 30) самих ДНК-копий протоонкогена c-Ha-ras.
Мутация протоонкогенов. Мутация протоонкогенов может возникать под действием химических, физических и биологических факторов. Поскольку сам протоонкоген исключительно мал, мутации может подвергаться крайне ограниченный его участок (один нуклеотид из тысячи). Тем не менее, если мутация состоялась, то она неизбежно ведет к качественным изменениям регулирующей функции онкогена и далее синтезу и накоплению под влиянием активированного онкогена онкобелка. Подтверждением роли мутаций в активации протоонкогенов могут служить эксперименты с микроинъекцией всего лишь единичной копии мутированного клеточного онкогена (например, копии активного c-Ha-ras), которой оказалось достаточной для формирования опухолевой трансформации клетки).
Инсерция и транспозиция. Экспериментальная активация клеточного протоонкогена возможна путем внесения в клеточный геном чужеродного (вирусного) генетического материала (см. выше, раздел «Включение промотора»). Активация возможна только в том случае, если «навязываемый» нуклеотиду материал встраивается в определенную позицию на ДНК вблизи протоонкогена. Только в этом случае активная вирусная «промоторная ДНК» превращает «молчащий» ген в действующий.
Трансдукция (См. выше).
В геноме человека имеется примерно 100 000 генов. Общее количество протоонкогенов и онкогенов среди них едва ли превышает 100. К 1994 году обнаружено 67 с-онкогенов (данные таблицы 3-6). Один и тот же c-онкоген может «работать» в разных опухолях и у разных видов животных. Так, клеточный онкоген N-ras активен в культурах клеток рака легкого, толстой кишки, фибросаркомы, рака печени, нейробластомы и промиелоцитарного лейкоза. С другой стороны, в клетках одной и той же опухоли могут проявлять активность несколько разных с-онкогенов. Например, в клетках саркомы Юнга обнаружены активные с-онкогены myb (миелобластоз), mye (миелоцитоз), fes (саркомы кошек). Это свидетельствует о том, что каждый, а не какой-то определенный с-онкоген может вызвать опухолевую трансформацию в любой нормальной клетке. Кроме того, для обеспечения опухолевой трансформации необходимо действие не одного единственного онкогена, а, по крайней мере, двух или, возможно, большего числа онкогенов.
В настоящее время уже ни у кого не вызывает сомнений, что клеточные протоонкогены представляют собой набор генов, регулирующих рост и дифференцировку клеток. C-онкогены кодируют синтез факторов роста, которые стимулируют пролиферацию. Наоборот, гены, вызывающие супрессию опухолевого роста, кодируют синтез белков, которые нейтрализуют действие ростовых сигналов. Мутации или активация онкогенов, кодирующих синтез ростовых факторов, и инактивация генов супрессии опухоли, приводят к опухолевой трансформации, которая выражается в экспрессии с-онкогенов и генов супрессии опухоли. Так, установлено, что онкогены, вовлекаемые в карциному молочной железы, кодируют выработку многих онкобелков со свойствами факторов роста: int-2, bst, bcl-1 (факторы роста), erb-2, erb-A, erb-B3 (рецепторы факторов роста), ras (преобразователи сигналов), myc, mys, fos (ядерные регуляторы), а гены-супрессоры опухолевого роста, кодирующие выработку соответствующих белков-супрессоров - rb-1 и p53.
В целом онкобелки, или раковые белки, синтезируются под влиянием активных с-онкогенов клетками, трансформированными в опухолевые. С помощью онкогенов измененная (опухолевая) генетическая программа реализуется в биологические признаки опухоли, или атипизмы. Выше указывалось, что образование онкобелков в следовых количествах в нормальных клетках кодируется протоонкогенами. Пока неизвестно, какую физиологическую роль играют протоонкогены и кодируемые ими белки в нормальных клетках. Считают, что они берут на себя функцию факторов роста, колониенстимулирующих факторов или подобные им функции.
Наряду со сложными взаимодействиями с-онкогенов и генов-супрессоров опухолевого роста практически при любом бластомном процессе обнаруживаются другие генетические аномалии. Среди них называют делеции (потеря участка хромосомы или хроматиды), транслока-ции, инверсии генов, моносомии, трисомии специфических хромосом. Так как такие аномалии хромосом встречаются при определенном типе опухолей, предполагается, что они вовлекаются в канцерогенеза. Например, колоректальная карцинома сопровождается экспрессией активных с-онкогенов (ras, mys, src), инактивацией или делециями генов-супрессоров опухолевого роста, а также делециями хромосом 1 и 22. Перечень некоторых хромосомных хромосомных аномалий представлен в табл. 3-7.
Научная электронная библиотека

Представления о молекулярно-клеточных механизмах онкогенной трансформации клеток претерпели значительную эволюцию на протяжении XX века и до настоящего времени [18, 20, 25, 32, 34].
Как указывалось выше, инициирующими этиологическими факторами малигнизации клетки являются разнообразные по природе группы канцерогенов химической, физической, биологической природы, в том числе вирусы, гормоны и генотоксические продукты их метаболизма [13, 26, 63].
Естественно, что при чрезвычайной гетерогенности этиологических факторов неоплазий не могла сформироваться достаточно быстро доминирующая концепция механизмов развития онкогенной трансформации клеток, их активации или промоции опухолевого роста с последующей опухолевой прогрессией. В ранних концепциях канцерогенеза делали акцент на эпигеномных механизмах развития неоплазий, и, безусловно, ряд положений этого направления носит не только исторический характер, но может быть в определенной степени ассоциирован с современными вирусо-генетической и онкогенной теориями канцерогенеза.
Согласно данным ряда исследователей, первичное изменение свойств цитоплазматической мембраны под влиянием канцерогенных углеводородов, онкогенных вирусов является одним из пусковых механизмов последующего изменения генетического аппарата и нарушений регуляции их митотического цикла [108]. Эта концепция канцерогенеза была актуальна в период обнаружения отсутствия контактного ингибирования опухолевых клеток в монослойной культуре.
Как оказалось далее, в механизмах контактного ингибирования клеток важная роль отводится активации мембранной аденилциклазы и увеличению уровня цАМФ, тормозящего митотическую активность клеток. Понижение концентрации цАМФ в мембранах клеток под влиянием различных канцерогенов ведет к неконтролируемой митотической активности. Эта точка зрения имела определенную значимость в понимании пусковых механизмов канцерогенеза, поскольку для многих гормонов, регулирующих метаболизм клеток, их митотическую активность, характерен преимущественно мембранный тип рецепции (АКТГ, СТГ, инсулин, пролактин и др.).
Практически одновременно с мембранной концепцией канцерогенеза создавались митохондриальная и лизосомальная теории развития неоплазий, согласно которым актомиозиновый белок митохондриальных мембран оказывается аномальным у малигнизированных клеток и утрачивает чувствительность к регулирующим влияниям АТФ; при этом гликолитическая реакция опухолевой клетки стимулируется митохондриальными факторами, поступающими постоянно в гиалоплазму, а возрастание концентрации АТФ не подавляет этот процесс.
Со временем митохондриальная теория канцерогенеза утратила свою актуальность, однако факт чрезмерной интенсификации гликолитических реакций, даже в условиях достаточной оксигенации опухолевых клеток, наличие обратного эффекта Пастера остаются неоспоримыми и характерными признаками метаболического атипизма опухолевых клеток. Наименее веской оказалась теория так называемого «лизосомального» канцерогенеза, согласно которой канцерогены вызывают лабилизацию мембран лизосом, активизацию и выход в цитоплазму гидролаз, в частности, ДНК-азы, обеспечивающей разрыв двойной связи ДНК и развитие опухолевой трансформации клеток. Однако, как известно, лизосомы – очень реактогенные субклеточные образования, проницаемость мембран которых резко возрастает под влиянием различных патогенных факторов экзогенного и эндогенного происхождения, далеко не всегда являющихся канцерогенами [32].
Одним из классических признаков неоплазий является нарушение регуляции дифференцировки и митотической активности клеток, в связи с чем указанная проблема затрагивается в той или иной форме в разных концепциях [1]. Однако до настоящего времени одной из ведущих концепций канцерогенеза является мутационная теория, согласно которой все канцерогены обладают мутагенной активностью, хотя не все мутагены являются канцерогенами.
Практически все изученные канцерогены индуцируют разрывы фосфодиэстеразных связей в молекуле ДНК. Вначале канцерогены интенсивно связываются с ДНК чувствительных клеток. Обнаружена прямая корреляция между чувствительностью животных и их органов к малигнизирующему действию веществ и степенью их связывания с ДНК [42].
Показано, что многие химические канцерогены способны к интеркаляции между основаниями ДНК с последующим сдвигом «рамки считывания» генетической информации. Установлено, что канцерогены различных классов взаимодействуют активно с нуклеиновыми основаниями ДНК; при этом ослабляется связь основания с сахарами, возникают гидролиз, денатурация ДНК. Горячими точками при индукции канцерогенами мутации сдвига «рамки считывания» являются полипуриновые участки ДНК. Возникновение повреждения под влиянием химических канцерогенов (полициклических углеводородов, ароматических аминов и амидов, алкилирующих соединений) может индуцировать процесс генетической рекомбинации, конверсии генов [66].
В последующие годы важная роль в развитии онкогенной трансформации клеток и опухолевой прогрессии отведена свободным радикалам. Учитывая значимость индукции избыточных концентраций свободных радикалов в канцерогенезе, необходимо прежде всего остановиться на активации процессов липопероксидации, инициируемой активными формами кислорода (АФК) и в то же время являющейся источником образования значительного количества вторичных эндогенных свободных радикалов [7, 8].
Как известно, активные формы кислорода вступают во взаимодействие с полиненасыщенными жирными кислотами (ПНЖК): линолиевой, линоленовой, арахидоновой – важнейшими компонентами фосфолипидов биологических мембран. Отрыв водорода от молекулы ПНЖК при участии АФК приводит к перемещению двойных связей с образованием гидроперекисей диеновых коньюгатов, которые затем метаболизируются во вторичные (малоновый диальдегид) и третичные продукты липопероксидации [66]. Перекисное окисление липидов затрагивает прежде всего фосфолипиды цитоплазматических мембран клеток, нарушая при этом энергозависимый трансмембранный перенос субстратов, процессы межклеточного взаимодействия. Биологическая активность АФК связана с синтезом простагландинов, лейкотриенов окислительной модификацией белков, нуклеиновых кислот, липидов. Одним из проявлений окислительной модификации белка является инактивация около 240 ферментов, в частности, СОД, ацетил-КоА-гидролазы, каталазы, миелопероксидазы, цитохрома Р450 [22, 66].
Дезинтеграция белка в основном возникает под влиянием гидроксильного радикала, образующегося в организме в процессе реакции взаимодействия супероксида и перекиси водорода с металлами переменной валентности. Объектами окисления в молекуле ДНК под влиянием гидроксильного радикала являются углеводные компоненты, фосфатные группировки, азотистые основания. Наиболее чувствительным к окислительной деструкции азотистым основанием является гуанин, модифицированные формы которого составляют 45 % от общего количества окисленных оснований [83, 95].
Установлено, что чувствительность к фрагментации сахарно-фосфатного остатка ДНК под влиянием АФК оказалось более высокой, чем полипептидного остова белково-пептидных субстанций. Гидроксильный радикал, действуя на ДНК, может отрывать атом водорода от дезоксирибозофосфата, что ведет к его расщеплению и освобождению азотистых оснований. При этом образуются высокотоксичные производные альдегиды.
Данные, опубликованные в последние годы, убедительно свидетельствуют о том, что активные формы кислорода, оксид азота и его производные в сочетании с инфекционными патогенными факторами, бактериями и вирусами, являются ключевыми факторами канцерогенеза [2, 35, 36].
Детальный обзор литературы по этому вопросу приведен в работе Х. Маеда, Т. Акаике (1998). Кислородные радикалы, а также оксид азота могут повреждать ДНК, вызывая мутацию. Мутагенный и канцерогенный эффекты указанных соединений резко возрастают при одномоментной, избыточной продукции, сопровождающейся их взаимодействием с образованием пероксинитрита. Последний участвует в различных внутриклеточных метаболических процессах: нитровании остатков тирозина в белках, подавлении транспорта электронов в митохондриях, в окислении тиоловых соединений. Пероксинитрит является ДНК-расщепляющим агентом. Вышеуказанные химические реакции с участием пероксинитрита могут инициировать апоптоз, мутации, онкогенную трансформацию клеток.
Как указывалось выше, в механизмах индукции канцерогенеза важная роль отводится онкогенным ДНК- и РНК-содержащим вирусам, способным инкорпорировать свою ДНК или ДНК-копию в геном хозяина с последующей возможной онкогенной трансформацией клетки в случае экспрессии протоонкогенов.
Установлено, что РНК-содержащие онкогенные вирусы являются членами семейства ретровирусов, характеризуются наличием липидной оболочки и двух односпиральных РНК, фермента РНК-зависимой ДНК-полимеразы, необходимой для репродукции вируса. Наличие этого фермента обеспечивает обратную транскрипцию вирусной РНК- в ДНК-копию, интегрирующую с геномом клетки [71].
Группа РНК-содержащих вирусов включает следующие разновидности: непатогенную для человека группу вирусов (род А); медленно трансформирующийся вирус гормонзависимой карциномы молочной железы морских свинок и, возможно, человека (род В); дефектные быстро трансформирующиеся и недефектные медленно трансформирующиеся вирусы (род С); род Д – включает вирусы приматов и вирус перевиваемых раковых клеток человека.
ДНК-содержащие онкогенные вирусы подразделяются на следующие семейства:
1. Семейство Poxviridae, содержит, в частности, вирус контагиозного моллюска человека.
2. Семейство Herpes viridae, к которому относится вирус Эпштейн-Барра человеа, вызывающий лимфому Беркитта, цитомегаловирус человека – тип 5.
3. Семейство Adenoviridae – представителями которого являются аденовирусы человека.
4. Семейство Papovaviridae, представителями которого являются вирусы папилломы крыс, хомяков, обезьян, человека.
ДНК-содержащие вирусы внедряют свою ДНК в геном хозяина при участии ферментов эндонуклеаз и липаз, а за счет наличия генов – промоторов – вирусы инициируют транскрипцию генов, следующих за ДНК-вирусами. Последствия внедрения ДНК-вирусов в геном хозяина зависят от зоны инкорнации: интронов, экзонов, протоонкогенов, антионкогенов. Если ДНК-содержащие вирусы встраивают в геном хозяина клетки регуляторы экспрессии протоонкогенов, возможна малигнизация клетки [54].
Механизмы онкогенной трансформации клеток под влиянием ДНК-содержащих вирусов могут быть весьма разнообразны: за счет индукции ранних онкобелков, так называемых Т-антигенов, усиления экспрессии рецепторов экзогенных ростовых факторов. Большие и средние Т-белки ряда ДНК-содержащих вирусов выключают контактное ингибирование пролиферации клеток, препятствуют действию антионкогена р53.
Как известно, вирусо-генетическая теория Л.А. Зильбера явилась основной для формирования современной онкогенной теории канцерогенеза. На смену вирусогенетической теории канцерогенеза пришли теории онкогенов, протоонкогенов и антионкогенов [30, 31, 65, 120].
В настоящее время, очевидно, что в опухолевой трансформации клеток, возникающей под влиянием различных индукторов канцерогенеза, принципиально участвуют следующие категории генов:
1. Онкогены- стимуляторы функций.
2. Гены роста и пролиферации клеток (Myc, Ras, Los, ABL и другие).
3. Антионкогены (потеря функции).
4. Гены, отвечающие за программированную смерть клетки (апоптоз):
– отменяющие программированную смерть: Bcl-2 (стимуляция функций);
– гены смерти клеток – р53 (потеря функции).
Онкогены как специфический химический материал, кодирующий информацию об определенном химическом продукте, впервые были идентифицированы в составе ретровирусов. Геном типичного не трансформирующего ретровируса представляет собой две молекулы односпиральной РНК. Основные гены вируса относятся к трем регионам: gag кодирует структурные белки вирион частицы, env– белки оболочки вириона, ген pol – несет информацию об обратной транскрипции. Последний обеспечивает образование ДНК- копии на матрице РНК-вируса.
Согласно гипотезе онкогенов, гены ретровирусов, попавшие в геном человека в процессе эволюции, переходят по наследству в ряде поколений, проявляют себя в раннем онтогенезе, а затем подавляются внутриклеточными репрессорами. С возрастом под влиянием различных канцерогенов физической, химической, биологической природы возникают экспрессия вирусных онкогенов и усиление продукции ими онкобелков, ответственных за малигнизацию клетки. Онкогенные свойства нетрансформирующих ретровирусов обусловлены наличием в их геноме V-онкогенов, причем большинство из 50 V-онкогенов имеют клеточные прототипы – С-протоонкогены.
Высказывается мысль, что ретровирусы не только могут вносить в определенные позиции клеточного генома V-онкогены, но и способны быть промоторами для усиленной экспрессии протоонкогенов клеток. Считается, что в ходе совместной эволюции ретровирусов и клеток происходят захват клеточных протонкогенов вирусами и их перенос [24].
Развитие теории онкогенов нашло отражение в концепции Темина (1972) о протовирусах, протоонкогенах, согласно которой предсуществующий аналог вируса не является результатом инфекции, а нормальным клеточным геном, необходимым для роста и онтогенеза клеток, причем нормальные клетки не содержат вирусных онкогенов, но зависят от контролируемой экспрессии их клеточных аналогов.
В механизмах развития неоплазий онкогенные ретровирусы играют неоднозначную роль: различают быстро- и медленно-трансформирующие вирусы. Быстротрансформирующие вирусы дефектны по структуре, утратили часть своих поздних репликативных генов и приобрели взамен видоизмененные клеточные гены-V-онкогены, которые и вызывают неопластическую трансформацию при повторной интеграции в клеточный геном. Для полного цикла репликации этим вирусом требуются вирусы-помощники. Клеточные протоонкогены являются прототипами V-онкогенов, консервативными регуляторами клеточной дифференцировки.
Встраивание быстро-трансформирующего реторовируса может либо привести к экспрессии в клетке V-онкогена, либо вирусные промоторы и энхансеры встраиваются рядом с протоонкогенами клетки, вызывая их экспрессию.
Медленно-трансформирующие ретровирусы вызывают в эксперименте рак молочной железы и хронические лейкозы; они способны самостоятельно реплицироваться в клетки, не содержат V-онкогенов, способны к «вставочному» мутагенезу. При этом возникает гиперэкспрессия клеточного протоонкогена.
Таким образом, встраивание ретровирусов в геном клетки приводит к гиперэкспрессии протоонкогенов, переход их в онкогены с последующей малигнизацией клетки [20, 23, 30, 64].
Что касается механизмов индукции неоплазий химическими канцерогенами с точки зрения современных теорий канцерогенеза – протоонкогенов, онкогенов, антионкогенов, то необходимо остановиться на анализе лишь некоторых работ, посвященных данной проблеме.
Как известно, химические канцерогены, подобно биологическим, способны вызывать развитие мутаций и активацию протоонкогенов [25, 64]. Под влиянием химических канцерогенов возможна онкогенная трансформация в процессе амплификации ДНК. Установлено, что амплификация гена резистентности на фоне воздействия цитостатиков нередко возникает при раке кишечника и является причиной устойчивости неоплазий к химиотерапии. При ряде онкологических заболеваний желудочно-кишечного тракта возникает амплификация онкогенов erbB2, mys, SRS. Индукция развития опухолей нитрозмочевиной связана с амплификацией и активацией N-ras; в опухолях, индуцированных гамма-облучением, активен Ras-H. В ходе химического канцерогенеза отмечено гипометилирование протоонкогена Ras-H, приводящего к развитию генной мутации.
В опухолях, индуцированных химическими канцерогенами, отмечены транскрипции ряда других онкогенов (c-ras и c-mys), связанные с гипометилированием протоонкогена либо его амплификацией. В ходе химического канцерогенеза нарушается зависимость экспрессии c-mys (но не c-ras) от клеточного цикла. Таким образом, многие химические соединения или физические воздействия, а также вирусы могут вызывать мутации ДНК, не летальные для клеток и провоцирующие экспрессию протоонкогенов или депрессию антипротоонкогенов [108]. Последнее приводит к трансформации нормальной клетки в опухолевую.
Эпигенетический механизм канцерогенеза связан с нарушением регуляции клеточного роста, функции клетки и экспрессии генов без повреждения генома. При эпигенетическом канцерогенном эффекте эндогенных или экзогенных канцерогенных факторов возникает инактивация белков-продуктов антипротоонкогенов или активация пострецепторных передатчиков ростовых факторов. Такое воздействие, как правило, не вызывает неоплазии, но усиливает ростовые эффекты, способствует пролиферации мутантного клона и формированию распознаваемой неоплазии. Эффект канцерогенов-мутагенов называют инициирующим, а коканцерогенов – активирующим.
Таким образом, в настоящее время очевидны следующие механизмы активации протоонкогенов:
1) амплификация протоонкогенов, в результате чего резко возрастает их общая активность, что может привести к малигнизации клетки;
2) мутации протоонкогенов, приводящие к их активации, и ингибиция антипротоонкогенов;
3) транслокация протоонкогенов в локус с функционирующим промотором;
4) аддукция промотора рядом с протоонкогеном. В качестве промотора могут выступать ДНК-копии определенных участков онкорнавирусов, а также мобильные генетические структуры, способные перемещаться и встраиваться в различные участки генома.
В геноме человека предполагается наличие около 100 протоонкогенов, выполняющих следующие функции:
1) кодирование ростовых факторов, их рецепторов и пострецепторных передатчиков;
2) кодирование блокаторов запрограммированной гибели клеток, контактного ингибирования пролиферации.
Трансформация протоонкогенов в онкогены приводит к их экспрессии и синтезу онкобелков. При этом онкобелки продуцируются перманентно в увеличенном количестве или в качественно измененном состоянии.
Ниже представлены несколько групп протоонкогенов, антионкогены, и кодируемые ими белки [30, 31, 32].
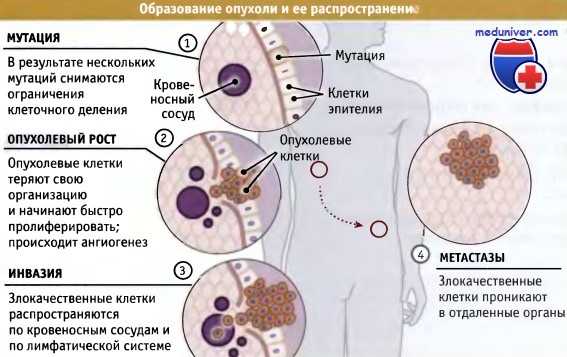
Механизм активации онкогенов и протоонкогенов клетки
• Опухоль развивается из одной мутировавшей клетки и затем дает метастазы
• Опухоли имеют клональное происхождение
• Опухоли классифицируются по клеточному типу
Рак представляет собой болезнь клеток, которые развиваются в организме в неправильное время и в несоответствующем месте. Когда в клетках происходят мутации, нарушающие регуляцию деления, они размножаются, образуя массы, которые называются опухоли. Опухоли незлокачественного происхождения (доброкачественные) не проявляют инвазивности и не влияют на остальные ткани, в то время как раковые опухоли (злокачественные) проявляют деструктивные свойства.
Опухолевый рост сопровождается ростом новых кровеносных сосудов, питающих опухоль; этот процесс называется ангиогенез. Наконец, злокачественные клетки могут покидать свою первоначальную локализацию и перемещаться на новые места в организме. Этот процесс называется метастазирование. Перечисленные события схематически представлены на рисунке ниже.
Вся масса клеток опухоли представляет собой моноклональный вырост, в котором все клетки являются потомками одной клетки-предшественника. Эта клетка-предшественник сохранила исходное повреждение, в результате которого она вступила на путь аберрантного роста и стала злокачественной. Хотя повреждение могло произойти за десятки лет до момента обнаружения опухоли, мы предполагаем, что потомки клетки-предшественника продолжают обнаруживать многие полученные от нее аберрации.
Таким образом, если мы будем учитывать характер аберраций в клетке-предшественнике, мы можем многое выяснить о свойствах ее потомков и, в свою очередь, прогнозировать свойства опухоли как целого. С этой точки зрения, рак представляется заболеванием клеток, и многие, если не все, свойства распространенных опухолей можно прогнозировать на основании свойств индивидуальных составляющих их клеток.
Рак может развиваться как в виде локальной опухоли, так и давать обширные метастазы.
На схеме показаны основные этапы опухолевого роста эпителиальных клеток в результате возникновения в них мутаций.
В организме человека клетки многих различных типов могут образовывать опухоли. При этом опухоль каждого типа характеризуется характерными гистологическими (т. е. микроскопическими) чертами и биологическими свойствами. Поэтому раковые опухоли могут развиваться более чем в 100 различных участках тела и обнаруживают широкий спектр свойств.
Даже в пределах опухолей одного типа наблюдается большая вариабельность гистологических и биохимических свойств клеток. Тем не менее, хотя специфика клеток может определять свойства опухоли (например, по сравнению с базальноклеточными карциномами, меланомы более инвазивны), все раковые клетки обладают такими общими фундаментальными биохимическими свойствами, как неконтролируемый рост и деление.
Опухоли классифицируются на четыре основные группы, которые определяются типом клеток, из которых происходят эти различные неоплазии или растущие массы аномальных клеток. Наиболее часто встречающиеся опухоли человека представлены карциномами, которые образуются при трансформации эпителиальных клеток, выстилающих полости и поверхности различных органов. К числу распространенных относятся карциномы легкого, кишки, молочной железы, предстательной железы, желудка, поджелудочной железы и кожи.
Несколько реже развиваются саркомы, которые возникают из мезенхимальной ткани, образующейся из фибробластов и близких по типу клеток; к этой же группе относятся опухоли костей и мышц. Частыми мишенями при канцерогенезе служат органы гемопоэза (кроветворения), и злокачественная трансформация соответствующих клеток приводит к развитию лейкозов, лимфом, миелом и близких заболеваний. К четвертой группе относятся опухоли из нейроэктодермальных клеток. Они включают нейробластомы, глиобластомы, нейромы, нейрофибросаркомы и меланомы. В таблице ниже представлена частота заболевания различными формами рака в США в 2017 г.
Онкология быстро развивается от простого описания различных и как будто бы не связанных между собой феноменов в стройную систему, в которой свойства клеток и опухолей можно выразить в рамках нескольких основных характеристик. В настоящем разделе описаны эти характеристики и то, каким образом с их помощью можно объяснить сложный и изменчивый характер опухолей человека.

Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
• Доминантно-негативные мутации могут активировать протоонкогены
• Гиперэкспрессия протоонкогенов может вызвать образование опухолей
• При транслокации могут возникнуть гибридные белки, проявляющие онкогенные свойства
Большинство опухолей человека развиваются не в результате инфекций, вызванных ретровирусами или ДНК-содержащими вирусами. Это поднимает вопрос о том, каким образом раковая клетка человека получает онкогены и утрачивает функции генов-супрессоров опухоли.
В 1979 г. было показано, что ДНК, выделенная из клеток опухоли мышей, развившейся в результате воздействия химического канцерогена, может быть перенесена в здоровые клетки (эта процедура называется трансфекция), в результате чего клетки-реципиенты трансформируются. Это свидетельствует о том, что химически трансформированные клетки содержат онкогены, которые ведут себя так же, как онкогены ДНК-или РНК-содержащих онкогенных вирусов.
Однако эти химически трансформированные клетки не имели отношения к процессу вирусной инфекции. Поэтому было высказано предположение, которое в течение последующих лет подтвердилось: трансформирующие онкогены, находящиеся в химически трансформированных клетках, представляли собой мутантные варианты нормальных клеточных протоонкогенов. Эти мутантные варианты образовались в результате действия химических канцерогенов.
Фактически онкоген, активируемый химическими канцерогенами, близок к Ki-ras онкогену, характерному для вируса крысиной саркомы Кирстена, представляющего собой мышиный ретровирус. Аналогичным образом, онкоген, обнаруженный при трансфекции ДНК карциномы мочевого пузыря человека, оказался идентичным онкогену На-ras, присутствующему в вирусе крысиной саркомы Харви. Вскоре в различных опухолях кроветворной системы человека также была обнаружена измененная форма онкогена myc, который первоначально был найден в геноме птичьего миелоцитоматоза.

Активация онкогенов может происходить за счет количественных и качественных изменений белков.
Все эти данные позволили прийти к очевидному в настоящее время выводу: активация протоонкогенов с образованием онкогенов может происходить по альтернативным механизмам. У некоторых животных активация происходит при включении протоонкогенов в геном ретровируса. В случае опухолей невирусного происхождения, например опухолей человека, те же гены могут активироваться за счет мутаций, которые изменяют нормальные гены, входящие в состав клеточных хромосом.
В клетках человека различные мутации служат причиной активации протоонкогенов в онкогены. В случае онкогенов ras простая точечная мутация в области рамки считывания приводит к превращению протоонкогена в онкоген. Эта мутация действует как доминантно-негативная в том смысле, что при ней в течение длительного, а не кратковременного периода, Ras-белок остается в конфигурации стимулирующей рост. Для онкогена myc, по крайней мере, два механизма могут обусловливать его активацию в клетках опухолей человека.
Несколько механизмов, которые приводят к дополнительному образованию онкогена, влияют на степень его экспрессии и, тем самым, приводят к повышению уровня Myc белка. Так, в некоторых случаях копийность туе гена увеличивается за счет процесса его амплификации. В других случаях, за счет транслокации, myc ген сливается с другим геном, например с иммуноглобулиновым. Вследствие этого экспрессия myc гена, которая в норме очень низкая и хорошо контролируется, попадает под контроль чужого промотора транскрипции. Это приводит к тому, что экспрессия белка достигает стабильно высокого уровня и становится неконтролируемым событием.
Онкогены могут возникать при хромосомных транслокациях совсем по другому механизму. Хороший пример представляет филадельфийская хромосома, обнаруженная у многих больных хроническим миелолейкозом (CML). Эта хромосома является результатом реципрокной транслокации, обменом хромосомным материалом между хромосомами 9 и 22. При транслокации происходит слияние генов Bcr и Abl и образуемся гибридная рамка считывания.
Особенность гибридного белка состоит в том, что он содержит N-концевой участок белка Bcr и большую часть белка Abl на С-концевом участке. Механизм, по которому этот гибридный белок приобретает онкогенные свойства, не вполне ясен. Впрочем известно, что белок АЫ кодирует сигнальную молекулу, которая по свойствам напоминает Src белок вируса саркомы Рауса. На рисунке ниже показаны различные пути возникновения доминантно-негативных мутаций в протоонкогенах.
Итак, как мы видели, трансформация клеток может происходить в результате как качественных (в случае ras и Bcr-Abl), так и количественных (в случае myc) изменений генов.
На пути к детальному каталогу раковых генов

Создание детального каталога раковых генов — важная задача, выполнение которой позволит подбирать оптимальную терапию онкологического заболевания для каждого пациента. Чтобы составить каталог раковых генов, мутирующих с высокой (>20%) и средней (2–20%) частотой, требуется проанализировать для каждого гена в среднем 2000 пар «опухоль — норма», то есть для 50 наиболее частых типов рака это примерно 100 000 пар. Сейчас это уже не является неразрешимой проблемой, так как за последние 10 лет стоимость секвенирования ДНК уменьшилась в миллион раз и будет уменьшаться еще.
В настоящее время рак занимает второе место среди причин смерти человека, уступая лишь сердечно-сосудистым заболеваниям (ежегодно в мире от рака умирает примерно 8 миллионов человек), а в некоторых развитых странах, например в Дании, рак уже вышел на первое место.
Рак — это сложное, динамически развивающееся заболевание, представленное более чем 200 известными типами и формами. Каждая из них требует индивидуального подхода, индивидуальной стратегии лечения. На генетическом уровне различные раки характеризуются различной «архитектурой» — наборами соматических мутаций, перестроек хромосом, а также эпигенетическими аномалиями, такими как изменение профиля метилирования генов. Следствием этих событий является изменение активности генов и(или) их продуктов.
Детальные сведения об аномалиях, связанных с возникновением и развитием раковых опухолей, требуются для диагностики и результативного лечения рака, для определения оптимальной терапии, а также для разработки новых противораковых средств. Объектами исследований являются аномалии молекулярной структуры ДНК, РНК, белков и эпигенетических аномалий (в частности, метилирования). Именно такой подход принят как генеральная стратегия, реализуемая несколькими национальными и международными консорциумами, которые объединяют десятки институтов, университетов и клиник. В работах участвуют сотни исследователей. Наибольшие успехи и наиболее ценные данные получены в результате поиска генов, связанных с инициацией, развитием и поддержанием злокачественной трансформации клеток.
Онкогены и антионкогены
В организме должен поддерживаться тонкий баланс между активностью генов и их продуктов, которые, с одной стороны, обеспечивают рост и деление клеток, а с другой стороны — предотвращают неограниченные рост и деление. Излишняя активность первых или подавление функции вторых приводят к неконтролируемому росту клеток, возникновению и развитию злокачественных неоплазий — раковых опухолей.
Связанные с раком гены можно разделить на два типа — онкогены и антионкогены (супрессоры опухолей), продукты которых могут, соответственно, стимулировать или подавлять образование и развитие опухолей. Особое место занимают микроРНК (miRNA) — короткие (в среднем ~22 нуклеотида) некодирующие РНК. К настоящему времени идентифицировано примерно 2000 различных микроРНК. Они способны подавлять трансляцию мРНК, считываемую с 30–60% генов человека. Некоторые микроРНК (oncomiR) способствуют злокачественной трансформации клеток, другие могут работать как антионкогены. В онкоген может превратиться нормальный ген — протоонкоген (рис. 1), который стимулирует рост клеток постоянно или на определенных стадиях развития организма. Трансформация протоонкогена в онкоген происходит в результате относительно незначительной модификации его естественной функции. Существуют следующие основные пути активации протоонкогена:
1) Мутация внутри протоонкогена или в его регуляторных элементах, которая меняет структуру белка и повышает активность кодируемого им белка (фермента) или усиливает экспрессию соответствующего гена.
2) Повышение концентрации белка из-за повышения его стабильности в клетке, увеличения периода полужизни и, соответственно, повышения активности.
3) Дупликация гена (увеличение количества копий), в результате чего повышается концентрация белка в клетке.
4) Транслокация гена, которая вызывает усиление его экспрессии или возникновение агрессивного гибридного гена.
Онкогенами являются, например, гены семейства Ras (сокр. от ‘Rat sarcoma’) — ГТФазы, участвующие в передаче сигналов, стимулирующих деление клеток.
Функция антионкогенов противоположна функции протоонкогенов. Антионкогены контролируют различные процессы, препятствующие злокачественной трансформации клеток:
1) Подавление избыточной экспрессии генов, обеспечивающих пролиферацию клеток.
2) Осуществление репарации ДНК (повреждения ДНК при подавлении репарации усиливают мутагенез и, как следствие, активацию протоонкогенов и инактивацию антионкогенов).
3) Координация пролиферации клеток с репарацией ДНК. Если репарация ДНК подавлена, они тормозят деление клетки и иницииируют апоптоз.
4) Контроль адгезии и механизмов контактного торможения делящихся клеток.
В общем, антионкогены ставят барьер неограниченному росту клеток. Утрата функции антионкогена разрушает этот барьер. Самым известным и часто мутирующим в раковых опухолях многих типов является антионкоген ТР53. Продукт ТР53 — фосфопротеин регулирующий транскрипцию ряда различных генов. В нормальной клетке он неактивен. При чрезвычайных событиях он активируется и выполняет роль «стража генома», осуществляя различные противораковые функции (рис. 2):
1) Активация системы репарации ДНК.
2) Если ДНК повреждена, ТР53 задерживает митоз делящихся клеток, блокируя переход из G1-фазы в S-фазу и предоставляя системе репарации время устранить повреждения.
3) Если устранить повреждения ДНК не удается, ТР53 включает программу гибели клеток — апоптоз.

В случае, если рак вызван протоонкогеном, обычно достаточно активации этого протоонкогена на одной из двух парных клеточных хромосом. А вот если рак возник из-за утраты эффекта антионкогена, то, как правило, требуются мутации или потеря обеих его копий.
Сравнительное секвенирование
К настоящему времени идентифицировано примерно 300 онкогенов и супрессоров опухолей. Раковые гены ищут с помощью сравнительного секвенирования — то есть сравнивают последовательности нуклеотидов в ДНК опухолей и нормальных тканей и затем идентифицируют отсутствующие в ДНК нормальной ткани соматические мутации, которые встречаются чаще, чем просто случайные события.
Эта стратегия реализуется в несколько этапов. Во-первых, требуется получить образцы опухолевой ткани от пациентов с достоверно поставленным диагнозом и детально описанной картиной течения заболевания. При этом образцы должны быть, по возможности, свободны от нормальных клеток. Для сравнения используются пробы нормальной ткани или крови пациента. Из ткани опухоли и нормальной ткани выделяют ДНК и иссследуют их с помощью секвенирования. В последнее время секвенирование проводится с помощью платформ нового поколения, позволяющих быстро и сравнительно недорого полностью секвенировать геном человека. Результаты сравнительного секвенирования затем анализируются с помощью специально разработанных весьма сложных математических и биоинформатических методик (рис. 3).
Рис. 3. На целостность генома воздействуют внешние факторы, такие как ультрафиолетовое излучение, сигаретный дым или вирусные инфекции. Разные участки генома мутируют с различной частотой при различных формах рака. Результаты сравнения последовательности нуклеотидов в ДНК опухолей и нормальных тканей анализируются с помощью математических и биоинформатических методик, которые учитывают частоты и спектр мутаций, порядок, в котором происходит репликация сегментов ДНК по всей длине хромосомы, а также уровень экспрессии генов, — это позволяет найти гены, достоверно мутирующие при раке. Рисунок из статьи L. Ding и M. Wendl, 2013. Differences that matter in cancer genomics
Генеральной целью этих проектов является создание детального каталога аномалий структуры генома, связанных с инициацией, прогрессией и поддержанием онкологических новообразований. Такой каталог позволит не только получить много новых данных по молекулярной биологии рака, но и усовершенствовать методы диагностики, лечения и профилактики рака, определять новые мишени для разработки противораковых средств. Систематические исследования в этом направлении уже позволили идентифицировать много новых раковых генов и даже целые классы раковых генов.
«Атлас раковых геномов»
К настоящему времени уже проанализированы данные, полученные на больших выборках «опухоль — нормальная ткань» по более чем 30 формам рака. Наиболее заметных успехов добился реализуемый главным образом институтами и университетами США консорциум «Атлас раковых геномов» (The Cancer Genome Atlas) с многозначительной аббревиатурой TCGA — этими же буквами обозначают четыре нуклеотида, входящих в состав ДНК. Основанный в 2005 году, TCGA регулярно публикует в ведущих научных журналах результаты своих исследований. Рассказать обо всех публикациях консорциума здесь не представляется возможным. Приведем лишь результаты из последней статьи, посвященной плоскоклеточным ракам головы и шеи. Эта гетерогенная группа раков — шестая по частоте встречаемости и составляет ~5% всех случаев рака в мире.
В исследованиях участвовали 348 авторов. Было проанализировано 279 пар «опухоль — норма». Большинство опухолей, связанных с вирусами папилломы человека, имели мутации в спиральном домене онкогена PIK3CA. Обнаружены новые аномалии, включающие утрату TRAF3, амплификацию участвующего в контроле клеточного цикла гена E2F1. В опухолях, связанных с курением, почти всегда наблюдались инактивирующие мутации генов TP53 и CDKN2A, обнаруживались амплификации участков хромосом 3q26/28 и 11q13/22. Опухоли ротовой полости, сравнительно благоприятные с точки зрения возможности лечения и шансов на выздоровление, содержали активирующие мутации генов HRAS или PIK3CA в сочетании с инактивирующими мутациями генов CASP8, NOTCH1 и TP53. В случаях других подгрупп этого рака найдены инактивирующие мутации гена NSD1, продукт которого связан с перестройками хроматина, инактивирующие мутации генов AJUBA и FAT1, контролирующих ферменты сигнального пути Wnt, мутации, активирующие фактор оксидативного стресса NFE2L2.
«Умеренные» и «редкие» онкогены
Некоторые связанные с раком гены мутируют довольно часто — при многих или, по крайней мере, при нескольких типах рака. Поэтому неудивительно, что именно они (в частности, TP53) были охарактеризованы первыми. Но большинство раковых генов обнаруживаются с умеренной частотой (2–20%) или реже. Основные проблемы возникают при идентификации редких раковых генов. Так, недавнее исследование 183 аденокарцином легких показало, что у 15% пациентов не обнаруживается ни одной мутации в 10 известных для этого заболевания классах генов, а в 38% случаев идентифицировано три или менее мутаций (M. Imielinski et al., 2012. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing).
Вследствие повреждения систем репарации ДНК в раковых клетках в той или иной мере усиливается мутагенез. Частота этих индуцированных опухолью соматических мутаций для различных опухолей может различаться на несколько порядков. Поэтому при идентификации «умеренных» и особенно «редких» генов возникает существенная проблема: как отличить гены и мутации, связанные с раком, от фона, от многочисленных случайных мутаций, не имеющих отношения к раку?
Гарвардская группа под руководством Г. Гетца (G. Getz) разработала программный пакет MutSig (сокр. от ‘Mutation Significance’), анализирующий частоты и спектр мутаций в различных областях генома при различных формах рака, а также ряд других факторов, и позволяющий вычленить гены, достоверно мутирующие при раке.
Мутации, достоверно связанные с раком
В двух статьях (M. S. Lawrence et al., 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes и M. S. Lawrence et al., 2014. Discovery and saturation analysis of cancer genes across 21 tumour types) авторы изучили собранные из различных баз данных результаты анализа экзомов (кодирующих областей генов — экзонов с прилежащими к ним последовательностями ДНК-некодирующих промежутков — интронов) 4742 пар «опухоль — нормальная ткань», принадлежащих к ракам 21 различных типов. Количество образцов для отдельных типов варьировало от 35 до 892.
Были обнаружены 3 078 483 единичные замены нуклеотидов в опухолях по сравнению с нормальной тканью, 77 270 единичных делеций или вставок нуклеотидов, 29 837 ди-, три- или олигонуклеотидных делеций или вставок. Из единичных замен нуклеотидов подавляющее большинство (2 294 935) не изменяло кодирующие последовательности. Из остальных единичных замен 540 831 представляли собой так называемые миссенс-мутации (приводящие к заменам аминокислот в белках), 207 144 представляли собой синонимические (не приводяшие к заменам аминокислот) замены нуклеотидов, 46 264 — нонсенс-мутации (приводящие к досрочной терминации синтеза белка), 33 673 — мутации, нарушающие сплайсинг мРНК (сборку кодирующих последовательностей мРНК из соответствующих блоков). Данные по «глубине» секвенирования и чистоте образцов опухолей позволяют оценить чувствительность анализа величиной больше 90% (рис. 4). Частоты возникновения мутаций на единицу длины генома для различных типов рака различались более чем на 5 порядков (от 0,03 до 7000 на миллион нуклеотидов ДНК), сильно различались и спектры мутаций.

Рис. 4. Количество пар «опухоль — норма» (по оси ординат), необходимых для определения 90% генов, достоверно мутирующих при раке с вероятностью 90%. Оценки приведены в зависимости от типа опухоли, средней частоты фоновых мутаций (по оси абсцисс), превышения частоты мутаций раковых генов над фоном (цветные линии). Для большинства типов опухолей количество проанализированных проб еще недостаточно даже для того, чтобы детектировать гены, мутирующие с частотой над фоном 5% и менее. Черными точками показано количество уже проанализированных проб. Рисунок из статьи M. S. Lawrence et al., 2014. Discovery and saturation analysis of cancer genes across 21 tumour types
Мутации, достоверно связанные с раком, обнаруживались в 224 различных генах. Для различных типов рака число мутантных генов сильно варьировало (от 1 до 58). Для 7 типов оно было меньше 10, а для двух (раки молочной железы и эндометрия матки) — более 30. Только 22 гена оказались достоверно связанными с более чем тремя типами рака. Анализ позволил идентифицировать практически все ранее известные гены, связанные с канцерогенезом. Найдено также 33 гена, мутации в которых ранее не были проассоциированны с раком. Эти гены связаны с делением клеток, апоптозом, стабильностью генома, регуляцией активности хроматина, иммунным ответом, превращениями РНК и гомеостазом белков. Среди еще 81 гена также должны быть гены, связанные с раком.
На основании полученных результатов авторы произвели расчеты: сколько пар «опухоль — нормальная ткань» требуется проанализировать, чтобы найти достоверно мутирующие при раке гены в зависимости от типа рака, частоты мутаций на единицу длины генома при данном типе, частоты мутаций данного гена при данном типе рака. Расчеты показывают, что для 17 из 21 проанализированного типа рака данных еще недостаточно, чтобы выявить гены, мутирующие с частотой не выше 5% от фона. А для 7 типов — даже при частоте не выше 10%. Также определено, что для составления каталога раковых генов, перекрывающих 90% случаев заболевания, требуется проанализировать около 650 пар «опухоль — норма», если средняя частота мутаций составляет ~0,5 на миллион пар оснований ДНК (как при нейробластоме). Или даже около 5300 пар, если частота ~12,9 на миллион (как при меланоме). Всего же, чтобы для ~50 известных типов рака составить каталог соматических мутаций для генов, мутирующих с высокой (>20%) и средней (2–20%) частотой, требуется проанализировать в среднем 2000 пар «опухоль — норма», то есть всего примерно 100 000 пар.
В общем, создание детального каталога раковых генов — важная задача, выполнение которой позволит подбирать оптимальную терапию онкологического заболевания для каждого пациента: воздействия на определенные сигнальные пути или иные процессы, поврежденные в каждом конкретном случае. Такой каталог нужен также для выбора мишеней при разработке противораковых средств, создания новых экспериментальных моделей животных и линий клеток для исследования рака, испытания новых средств и методов лечения.
Источник: Cancer Genome Atlas Network. Collaborators (348). Comprehensive genomic characterization of head and neck squamous cell carcinomas // Nature. 2015. V. 517. P. 576–582.
См. также:
1) Marcin Imielinski et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing // Cell. 2012. V. 150. P. 1107–1120.
2) Michael S. Lawrence et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes // Nature. 2013. V. 499. P. 214–218.
3) Li Ding & Michael C. Wendl. Differences that matter in cancer genomics // Nat Biotechnol. 2013. V. 31. P. 892–893.
4) Michael S. Lawrence et al. Discovery and saturation analysis of cancer genes across 21 tumour types // Nature. 2014. V. 505. P. 495–501.
Читайте также: