Аплазия и гипоплазия мозжечка - клиника, диагностика
Обновлено: 17.05.2024
Аномалии формирования центральной нервной системы — отнюдь не редкость. Эта патология занимает второе место по частоте после врожденных дефектов развития сердца и сосудов и в большинстве случаев представлена гидроцефалией самого разного происхождения. Помимо тяжелых нарушений, сопровождающих патологическое развития мозга, пороки ЦНС несут высокий риск смертельного исхода, а по данным статистики они лидируют по числу смертей в младенческом возрасте.
Синдром Денди-Уокера — одна из разновидностей нарушения формирования головного мозга еще во время внутриутробного развития. И хотя частота его относительно невелика (всего 1 случай на 25-30 тысяч младенцев), диагностируется порок едва ли не у каждого десятого малыша с врожденной гидроцефалией, которая и служит одним из основных проявлений патологии.
Синдром Денди-Уокера — это порок задней черепной ямки, при этом основные структурные изменения касаются мозжечка, ликвороотводящих путей, четвертого желудочка мозга. Аномалия диагностируется во время беременности посредством ультразвукового осмотра, после чего женщине может быть предложено прерывание беременности по медицинским показаниям.
Конечно, любые отклонения в развитии плода — это всегда большой стресс и переживания для родителей, однако в случае врожденного порока мозга надеяться на чудо не приходится — прогноз серьезный, а смертность высока. Малыши с сочетанными пороками мозга и других органов погибают в раннем возрасте как от мозговой дисфункции, так и от присоединившейся инфекции.
Аномалия Денди-Уокера часто сочетается с другими нарушениями и генетическими заболеваниями, зачастую несовместимыми с жизнью — микроцефалия (недоразвитие полушарий мозга), мозговые грыжи. У младенца может быть диагностирован генетически обусловленный поликистоз почек, недоразвитие зрительных нервов и глазных яблок со слепотой, аномалии сердечно-сосудистой системы.
Все эти неблагоприятные факторы, возможность сочетанной патологии многих органов делают синдром Денди-Уокера серьезнейшей проблемой в случае, если малышу дадут возможность родиться. Лечение патологии, как правило, симптоматическое, направленное на поддержание главных систем жизнеобеспечения и борьбу с инфекционными осложнениями. В редких случаях применяют хирургическую операцию, которая лишь облегчает явления гидроцефалии, но не ликвидирует ее полностью.
Почему возникает синдром Денди-Уокера?

Причины аномалий развития задней черепной ямки до сих пор не выяснены, однако выделен ряд факторов, способствующих подобным врожденным порокам:
- Инфицирование во время беременности цитомегаловирусом, перенесенная краснуха;
- Употребление алкоголя, курение, наркомания во время беременности;
- Экстрагенитальная патология, особенно — сахарный диабет у будущей мамы.
Под действием перечисленных причин или среди полного благополучия может возникнуть спонтанная мутация в генах, предрасполагающая к нарушению развития мозга. Особенно высок риск пороков при действии неблагоприятных факторов во время первого триместра гестации, когда и происходит закладка основных структур центральной нервной системы.
В части случаев синдром Денди-Уокера носит наследственно-обусловленный характер, то есть возникает из-за дефекта генов и может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.
Что происходит с мозгом при синдроме Денди-Уокера?
Анатомически классический вариант мальформации задней черепной ямки включает:
- Гидроцефальный синдром разной степени выраженности;
- Кистозную полость в задней части черепа с расширенным четвертым желудочком мозга;
- Отсутствие или недоразвитие червя мозжечка, недоразвитие его полушарий.
Червь мозжечка — это структура, расположенная между его половинами и несущая в себе проводящие нервные волокна. При аномалии Денди-Уокера он может быть представлен небольшой щелью или широким пространством между гемисферами органа. При неполном отсутствии червя щелевидное расширение образуется лишь в нижней его части. На фоне патологии этого отдела наблюдается недостаточное развитие и мозжечковых полушарий.
Именно дефект мозжечкового червя в виде расщелины считается характерным признаком аномалии Денди-Уокера, позволяющим отличать ее от недоразвития на фоне других пороков мозга.

Киста четвертого мозгового желудочка может самопроизвольно вскрыться в 3-ий желудочек или субарахноидальное пространство. В этом случае симптомы окклюзии ликворных путей несколько ослабнут. Выраженность гидроцефального синдрома вариабельна — от небольшого расширения желудочковой системы до высокой степени окклюзионной гидроцефалии с отсутствием возможности для циркуляции ликвора.
Многие специалисты отмечают, что у большинства малышей с синдромом Денди-Уокера при рождении гидроцефалии как таковой нет, а формируется она и прогрессивно нарастает в течение первых нескольких месяцев жизни, поэтому факт отсутствия гидроцефального синдрома сразу после родов при наличии диагностированной внутриутробно патологии не является поводом для пересмотра диагноза и необоснованных надежд, с этим связанных.
Более, чем в половине случаев синдрома Денди-Уокера у детей помимо описанных структурных аномалий обнаруживаются и другие дефекты мозга — недоразвитие или отсутствие мозолистого тела, мозговые кисты, недоразвитие или отсутствие извилин, смещения серого вещества относительно его правильной локализации, что еще больше усугубляет течение и без того тяжелой патологии.
По данным МРТ было выделено несколько разновидностей синдрома Денди-Уокера:
- Классический тип аномалии — задняя черепная ямка расширена, четвертый желудочек кистозно изменен, червь мозжечка частично или полностью недоразвит, полушария его гипоплазированы, а намет находится выше, чем в норме, желудочковая система не сообщается с подпаутинным пространством, часто наблюдаются мозговые кисты и отсутствие мозолистого тела, практически у всех пациентов есть гидроцефалия, возможно сдавление стволовых структур. Порок проявляется клинически уже с рождения и имеет неблагоприятный прогноз.
- Вариант Денди-Уокера — морфологические признаки выражены меньше, чем при классической форме, гипоплазирован нижний отдел червя мозжечка, желудочки сообщаются с кистой и ликворными пространствами, обеспечивая отток ликвора, поэтому гидроцефалия наблюдается редко. Задняя черепная ямка имеет нормальные размеры, стволовые структуры не сдавливаются.
- Киста кармана Блейка — расширение желудочковой системы с гидроцефальным синдромом, киста расположена под или за мозжечком, червь развит относительно хорошо. Четвертый желудочек расширен, но не сообщается с затылочной ликворной цистерной.
- Mega cisterna magna — вариант очагового расширения подпаутинного пространства в задней и нижней частях задней ямки черепа с увеличением объема затылочной цистерны, которая сообщается с четвертым желудочком и субарахноидальным пространством.
Проявления заболевания
Симптоматика синдрома Денди-Уокера разнообразна. Возможны как практически нормальное развитие ребенка после рождения, так и грубые неврологические изменения, влекущие тяжелую инвалидность и даже смерть. По некоторым данным, нормальное развитие интеллекта бывает в половине случаев изолированного порока, возможно даже случайное обнаружение синдрома при обследовании взрослых.

дети с синдромом Денди-Уокера
Внутриутробное течение патологии определяется степенью поражения мозга, нарастанием гидроцефалии, наличием других пороков развития. Прогноз значительно хуже при диагностике синдрома до рождения. При глубоких нарушениях в формировании мозга на первый план среди других проявлений выступает гидроцефальный синдром:
- Увеличение диаметра головы;
- Выбухание родничка.
Увеличение диаметра черепа происходит, главным образом, за счет затылочной области, в которой образуется киста, вызывающая истончение и растяжение костной основы. При выраженной гидроцефалии голова малыша активно растет на протяжении первых двух месяцев, параллельно происходит расхождение швов между костями спереди или в заднем отделе. Кроме того, характерны:
- Повышение нервной возбудимости (рефлексов);
- Глазодвигательные расстройства — нистагм, косоглазие;
- Приступы остановки дыхания;
- Парез лицевого нерва.
Симптомы мозжечковых нарушений у новорожденных выявить невозможно, и даже тяжелый дефект формирования мозжечковых структур далеко не всегда вызывает значимые признаки атаксии (двигательных расстройств), которая регистрируется всего у трети пациентов.
Значительно чаще, нежели двигательные нарушения, возникают расстройства психической деятельности, интеллекта, которые проявляются на фоне общей двигательной «неловкости». У 25% больных с гипоплазированным мозжечком имеются признаки аутизма, в связи с чем специалисты пытаются найти взаимосвязь между изменениями мозжечка и аутизмом у детей.
Дети с гидроцефалией в раннем возрасте беспокойны, плохо спят, характерен монотонный крик, усиление рефлексов, плавающие движения глаз и их закатывание, выраженность сосудов роговицы, заметная подкожная венозная сеть по мере роста размеров головки. Спонтанная двигательная активность новорожденных может быть ослаблена, возможны судороги и тетрапарез из-за гипертонуса мышц.
В более старшем возрасте становится заметным отставание в психическом и интеллектуальном развитии, дети не могут обучаться, быстро устают, плохо усваивают новую информацию, что делает процесс адаптации крайне затруднительным. В тяжелых случаях обучение невозможно совсем, в связи с чем ребенок нуждается в постоянной посторонней помощи, уходе и рассмотрении вопроса об инвалидности.
Моторное развитие заметно замедлено. При тяжелых формах аномалии дети не могут своевременно научиться переворачиваться, ползать, садиться и ходить, не удерживают взгляд на игрушках, быстро устают и часто плачут. Возможны нарушения питания с гипотрофией, общее снижение иммунитета, частые инфекционные заболевания.
Сочетание порока нервной системы с другими аномалиями развития органов предрасполагает к серьезным осложнениям, в числе которых не только мозговая дисфункция, слабоумие, судорожный синдром, но и сердечная недостаточность, склонность к пневмониям при пороках сердца, хроническая почечная недостаточность и уремия при врожденном поликистозе, что усугубляет явления отека мозга и может послужить причиной гибели пациента.
При тяжелой окклюзионной гидроцефалии смерть может наступить в раннем младенчестве от отека головного мозга, фатальных аритмий, остановки дыхания на фоне компрессии стволовых структур, тяжелой пневмонии и других инфекционных осложнений.
У взрослых возможно постепенное нарастание гидроцефалии с краниалгиями, снижением памяти и внимания, раздражительностью, склонностью к депрессии, утренней тошнотой и рвотой на высоте головной боли. В тяжелых случаях бывает судорожный синдром. Возможны проблемы с координацией и выполнением мелких движений, неуверенность при ходьбе, зрительные расстройства.
Диагностика и лечение
Диагностика синдрома Денди-Уокера основывается на результатах ультразвукового осмотра, при этом важно обнаружить аномалию еще во время эмбрионального развития. УЗИ становится информативным после 18 недели гестации, но в некоторых случаях заподозрить патологию можно и раньше — уже на 14-15 неделях эмбрионального развития.

Диагностическими критериями при аномалии задней черепной ямки считаются:
- Наличие крупной кистозной полости, включающей четвертый мозговой желудочек, в задней части черепа;
- Отсутствие или аномальное развитие червя мозжечка;
- Гипоплазия мозжечковых гемисфер, наличие широкой щели между ними;
- Расширение желудочковой системы (гидроцефалия).
Для постановки диагноза синдрома Денди-Уокера необходимы:
- УЗИ (нейросонография);
- МРТ для определения анатомических особенностей четвертого желудочка мозга;
- Консультация офтальмолога;
- Осмотр нейрохирурга;
- УЗИ сердца для исключения врожденных аномалий;
- Консультация генетика и определение кариотипа при возможных генетических мутациях.
Лечение патологии определяется симптоматикой и тяжестью проявлений. Если гидроцефалии нет, а внутричерепное давление в пределах нормы, то оправдано динамическое наблюдение невролога, педиатра или нейрохирурга, каких-либо медикаментов не требуется.

шунтирование для нивелирования гидроцефалии
При нарастании гидроцефалии и внутричерепного давления показаны шунтирующие хирургические операции для отвода ликвора из черепа в грудную или брюшную полость. Медикаментозное лечение включает применение диуретиков (диакарб, маннитол), ноотропных средств (пирацетам, пантогам), антиконвульсантов (депакин).
В случае гипертонуса показаны физиотерапевтические и водные процедуры, массаж, специальные упражнения. Важен тщательный уход и постоянное наблюдение за малышом, создание спокойной обстановки при беспокойном поведении и нарушениях сна.
При тяжелых формах течения патологии с отставанием в интеллектуальном развитии детям показана работа с дефектологами-педагогами, психологом по индивидуальной программе, исключающей избыток информации и умственное перенапряжение.
Прогноз при синдроме Денди-Уокера зависит от ряда причин: времени установления диагноза, наличия других пороков и хромосомных болезней, степени окклюзии ликворных путей. Смертность и заболеваемость после рождения выше в тех случаях, когда аномалия сочетается с другими дефектами и обнаружена до рождения малыша.
Гидроцефалия и внутричерепная гипертензия — ключевые моменты в определении прогноза, которые влияют и на развитие пациента, и на продолжительность и качество его жизни. В случае изолированного поражения мозга без признаков гидроцефалии прогноз благоприятный. Ребенок может развиваться по возрасту, а иногда аномалия и вовсе выявляется у взрослых при обследовании по поводу других причин.
В связи с тем, что причины порока так и не выяснены, проводить специфическую профилактику не представляется возможным. Конечно, нужно соблюдать здоровый образ жизни, особенно, женщинам, планирующим беременность или уже забеременевшим с исключением вредных привычек, неблагоприятных влияний внешней среды. Важно своевременно выявить и пролечить цитомегаловирусную инфекцию, герпес, а в случае краснухи, которой женщина заболела при беременности, врачи предложат аборт по медицинским показаниям из-за высокого риска сочетанных пороков.
Решать вопрос о сохранении беременности в том случае, если синдром возник случайно, у плода абсолютно здоровой женщины, придется будущей маме и ее семье. Решение всегда дается сложно, но следует знать, что аномалия нервной системы и нормальное развитие и рост ребенка — скорее, исключение из правил.
В абсолютном большинстве случаев детям и родителям приходится бороться с гидроцефалией, зачастую требуется не одна дорогостоящая и сложная операция, тогда как ее эффективность и прогноз все равно могут оставаться сомнительными.
Видео: примеры детей с синдромом Денди-Уокера
Аномалии развития головного мозга ( Пороки развития головного мозга )
Аномалии развития головного мозга — это результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
МКБ-10

Общие сведения
Причины
Наиболее весомой причиной сбоев внутриутробного развития является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев. Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ.
Различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
Патогенез
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества.
Виды аномалий мозга
Анэнцефалия — отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле — пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия — уменьшение объема и массы головного мозга, обусловленное задержкой его развития. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия — увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия — характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия — отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия (гладкий мозг, лиссэнцефалия) — отставание развития извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия — укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия — поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия (ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии — скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Диагностика
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Лечение аномалий мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз и профилактика
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии. Эффективной мерой профилактики служит исключение эмбриотоксических и тератогенных влияний на женщину в период беременности. При планировании беременности будущим родителям следует избавиться от вредных привычек, пройти генетическое консультирование, обследование на наличие хронических инфекций.
Агенезия мозолистого тела
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.

Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».

Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
- Генетические аномалии. Повреждения мозолистого тела отмечаются при различных наследственных синдромах: Миллер-Дикера, Рубинштейна-Тауби, Доннаи-Кугана. Состояние входит в состав не менее 7 аутосомно-доминантных, 23 аутосомно-рецессивных, 12 Х-сцепленных врожденных заболеваний.
- Сосудистые нарушения. Причиной недоразвития мозолистого тела могут выступать артериовенозные мальформации или аневризмы, которые характеризуются отсутствием нормальной капиллярной сети. При этом возникает феномен обкрадывания, клетки МТ не получают должного количества кислорода, питательных веществ.
- Токсические влияния. Болезнь связана с действием тератогенных химических факторов: лекарственных препаратов, солей тяжелых металлов, пестицидов и бытовой химии. Негативное влияние на формирование ЦНС плода оказывает вдыхание табачного дыма (активное или пассивное курение) или прием беременной алкоголя во время гестации.
- Внутриутробные инфекции. Аномалии формирования неврологических структур, в том числе агенезия мозолистого тела, встречаются при проникновении возбудителей в организм плода на 2-3 месяце беременности. Нейротропные свойства демонстрируют герпетические инфекции, токсоплазмоз, цитомегаловирус.
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
- Агенезия. Закладка эмбрионального зачатка МТ отсутствует полностью.
- Аплазия. Эмбриональный зачаток мозолистого тела есть, но не развивается.
- Гипоплазия. МТ недостаточно развито из-за нарушений на одном из этапов эмбриогенеза: размеры и масса органа уменьшены, его функциональная активность снижена.
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
- КТ головного мозга. При компьютерной томографии определяются широко расставленные передние рога, высокое стояние третьего желудочка, параллельный ход медиальных стенок боковых желудочков. КТ производится в рамках постнатальной диагностики.
- МРТ головного мозга. Для максимально точной визуализации степени агенезии или гипоплазии мозолистого тела новорожденным выполняется магнитно-резонансная томография в трех плоскостях. По показаниям МРТ может рекомендоваться беременным женщинам для исключения несовместимых с жизнью сочетанных пороков ЦНС.
- Нейропсихологическое обследование. Для изучения когнитивных функций у детей применяется шкала интеллекта Векслера (WISC-Revised), адаптированное чтение и правописание (Schonnel Graded Reading and Spelling Tests), оценка вербальной беглости, тест контролируемых устных ассоциаций (Controlled Oral Word Association Test).
- Генетический анализ. Для подтверждения или исключения наследственных заболеваний, сопровождающихся агенезией мозолистого тела, показаны кариотипирование, секвенирование генома, проводимое как новорожденным, так и детям другого возраста. Исследования также проводят в антенатальном периоде для принятия решения о сохранении или прерывании беременности.
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
- Нейрологопедические программы. Занятия с детским логопедом проводятся для становления речевой функции, ликвидации проявлений дизартрии, улучшения артикуляции.
- Дефектологические программы. Помощь коррекционных педагогов требуется детям с интеллектуальными нарушениями, которые не могут проходить обучение в обычных классах.
- Нейроакустические программы. Формирование и гармонизация высших психических функций производятся с помощью звуковой терапии, музыкотерапии.
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
1. Эпилептические проявления когнитивные и аутистические расстройства у пациентов с агенезией мозолистого тела: результаты нейропсихологического тестирования/ О.А. Милованова, О.А. Комиссарова, Т.Ю. Тараканова, С.В. Бугрий// Эпилепсия и пароксизмальные состояния. — 2018. — №4.
2. Влияние особенностей строения мозолистого тела и доминирующего полушария на протекание психических процессов в юношеском возрасте/ У.С. Чернышова, Т.Ю. Хабарова, Д.А. Соколов// Центральный научный вестник. — 2016.
4. Молекулярная эмбриология: на пути каталогизации генов врожденных пороков развития головного мозга/ В.П. Пишак, М.А. Ризничук// Международный журнал педиатрии, акушерства и гинекологии. — 2014. — №5.
Гипоплазия левой позвоночной артерии: что это такое, описание симптомов, лечение, прогноз
Гипоплазия левой позвоночной артерии - это врожденное недоразвитие сосуда, суть которого - неадекватно малый просвет по сравнению с нормой. Компенсируется ситуация расширением позвоночной артерии с противоположенной стороны.
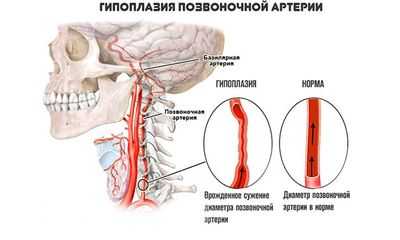
Заболевание не сопровождается какими-либо симптомами до определенного момента. Сразу нужно сказать, что право- или левосторонняя локализация — какой-либо роли это не играет. Симптоматика, прогнозы и методы терапии полностью совпадают. Равно как и причины возникновения, этиология. Однако слева дефект встречается почти втрое реже. Клиника отсутствует, пока нет еще большего сужения. На фоне, например, атеросклероза. Симптоматика соответствует общему и очаговому поражению головного мозга. Имеет неврологический характер. Терапия консервативная. По потребности назначается операция. Прогнозы в основном благоприятные. Зависит от течения ситуации.
Гипоплазия позвоночной артерии слева — врожденный порок анатомического развития. Как правило, это результат одного из двух процессов.
Спонтанное нарушение
Сердечнососудистая система формируется в первом триместре. В этот момент на женщине лежит колоссальная ответственность. Любой сбой может сказаться на состоянии будущего плода не известным образом. Патология закладывается именно в этот момент. Дают знать о себе перенесенные и вялотекущие инфекционные патологии, стрессы и физические перегрузки, особенно вредные привычки вроде курения и злоупотребления спиртным. В некоторых ситуациях фактор неочевиден: как мать ведет себя примерно, вынашивает плод, следуя всем рекомендациям.
Однако причина может быть в неблагоприятной экологии. Отсюда прямое влияние токсинов, радиационного фона на организм женщины и будущего плода.
Хромосомные мутации
Они почти не бывают спонтанными. Это результат либо несовместимости, либо влияния ряда факторов. Таких как ионизирующее излучения, поражение вирусами (особой опасностью обладают герпетические агенты, возбудители папилломатоза). Также возможны загрязнения атмосферы, воздействие соединений металлов. В такой ситуации гипоплазия оказывается далеко не «одинокой» проблемой. Параллельно возникает масса нарушений функционального и анатомического плана. Нередко эмбрион оказывается нежизнеспособен. Наступает выкидыш. Возможно и более позднее прерывание беременности.
Приобретенной гипоплазия не бывает. Как правило, просвет позвоночной артерии достаточен для обеспечения адекватного кровотока в церебральных структурах. Потому первые годы жизни, а то долгий период не омрачен плохим самочувствием или нарушениями работы головного мозга. Как только развивается атеросклероз или симптоматическое сужение сосуда, последствия могут быть катастрфоическими. Механизм двигается по типу ишемии вследствие нарушения гемодинамики, кровотока. Суженная артерия не может пропускать достаточное количество жидкой ткани. Страдает затылочная доля, мозжечок и вся экстрапирамидная система. Косвенно и иные области ЦНС.
При незначительном объеме нарушения проблема заметна, есть симптомы. Но критических состояний нет. Постепенно нарастают структурные изменения в мозгу. Расширяются желудочки. В конечном итоге наступает пик. Когда отклонение достигает определенного момента, развивается ишемический инсульт. Острое нарушение кровообращения с отмиранием нервных тканей и формированием стойкого неврологического дефицита. Состояние нередко становится причиной смерти.
Согласно статистике, 75% всех инсультов приходится именно на затылочную долю и мозжечок.
Потому нужно осознавать всю серьезность положения и проходить профилактические осмотры раз в 6 месяцев. По необходимости следовать рекомендациям врача.
Факторы развития патологического процесса всегда врожденные. Человек успевает получить проблемы, еще не родившись. Это может быть вина матери или стечение обстоятельств.
Среди возможных провокаторов:
- Курение в период вынашивания плода. Потребление спиртного в любых, даже самых малых количествах, тем более использование наркотиков.
- Прием препаратов, бесконтрольный или не по схеме.
- Частое нахождение на солнце.
- Физические перегрузки. Будущей матери полезно двигаться, но в меру и с умом.
- Стрессовые ситуации. Приводят к выбросу большого количества кортизола, адреналина. Также катехоламинов. Они могут сказаться на состоянии ребенка негативным образом.
- Влияние экологического фактора. Высокий радиационный фон, магнитное излечение свыше допустимой нормы, загрязненный воздух, вода, насыщенная ионами тяжелых металлов и ядовитых веществ.
- Неправильное питание. Отсутствие витаминов в рационе. Преобладание животного жира, фаст-фуда и различных неестественных добавок.
- Потребление кофеина. На период беременности от него лучше отказаться.
- Остро текущие инфекционные расстройства независимо от локализации. Но в особенности, затрагивающие репродуктивную систему. Также латентные их варианты, носительство.
- Нужно сказать, что ни один из названных факторов не дает стопроцентной гарантии гипоплазии у ребенка. Это всего лишь увеличивает риски. И чем больше таких моментов, тем выше вероятность.
Причины нужно рассматривать в системе.
Характерные симптомы
Симптомы данного недуга очень многообразны и могут значительно различаться у разных пациентов.

Вот несколько групп симптомов:
| Группа симптомов | Описание |
|---|---|
| Вертебральные | Боли в шейном отделе, затылке |
| Локальные | Боли с иррадиацией в голову при пальпации в точке позвоночной артерии - между поперечными отростками 1 и 2 шейных позвонков |
| Симптомы, связанные с ухудшением кровотока в вертебробазилярной системе или с раздражением симпатических волокон нервного сплетения вокруг позвоночной артерии | Повышение артериального давления, головная боль, зрительные расстройства, нарушение слуха, координации, неустойчивая походка, головокружения, нарушение чувствительности |
Характеристика проявлений болезни:
- Боли при патологии могут значительно различаться по интенсивности и другим характеристикам.
- Нередко пациенты ощущают пульсирующую или простреливающую боль с распространением от шеи и затылка до височно-лобных областей.
- Боли усиливаются при поворотах головы, по ночам и после пробуждения.
- Часто гипоплазия проявляется головокружениями, ощущением дезориентации, искажением восприятия положения тела в пространстве. Такие эпизоды нередко бывают связаны с наклонами головы, резкими движениями. Они могут приводить к пошатыванию или даже падению.
- Резкие приступы головокружения иногда сопровождаются потерей сознания, обмороками.
Кроме болевого синдрома при патологии могут возникать следующие нарушения:
- ухудшение зрения, боли в глазах, двоение в глазах, ощущение песка или мелькание мушек;
- ухудшение слуха, шум в ушах, нейросенсорная тугоухость, вестибулярные нарушения;
- проблемы со стороны сердечно-сосудистой системы;
- изменчивость настроения, угнетенное состояние;
- утомляемость, слабость;
- нарушение сна;
- метеочувствительность.
Артериальная гипертензия, приступы стенокардии не всегда являются прямым следствием аномалии позвоночных сосудов. Обычно сочетание кардиологической патологии с гипоплазией приводит к усугублению течения заболевания. При этом сниженный приток крови в вертебробазилярном бассейне провоцирует эпизоды ишемии миокарда и рост артериального давления. Гипоплазия правой позвоночной артерии повышает риск развития мозгового инсульта из-за нарушения кровотока в вертебробазилярной системе и за счет поражения сосудистой стенки в случае развития атеросклероза.
Проводится под контролем невролога, при необходимости привлекается профильный хирург. Обследование довольно простое, дает качественные результаты сразу, не считая сложных ситуаций. Среди мероприятий:
- Устный опрос пациента. Чтобы зафиксировать жалобы, составить перечень симптомов и выдвинуть гипотезы.
- Сбор анамнеза. Особенно врачей интересует течение беременности у матери, если такие данные имеются. В меньшей степени образ жизни, привычки, наследственный фактор.
- Допплерография сосудов шеи, головного мозга. Также дуплексное сканирование. Исследования, направленные на визуализацию тканей, оценку скорости кровотока и его качества. Определение нарушений, тяжести таковых, локализации. Возможно проведение с функциональными тестами. Поворотами головы и т.д. Это дает более полную картину.
- Ангиография. По необходимости. Для визуализации сосудов.
- Тот же эффект достигается посредством МРТ, только степень детализации в разы выше. Назначается в спорных случаях.
- Анализ крови общий и биохимия с расширенной картиной по липидному спектру. В рамках диагностики атеросклероза. Позволяет обнаружить избыток холестерина и других жирных соединений. Которые могут стать виновником болезни.
Этого достаточно. По необходимости показаны консультации прочих специалистов, но это уже частные предполагаемые случаи. Другие сценарии маловероятны.
Особенности лечения
В случае с гипоплазией сосудов полное излечение заболевания невозможно. Даже после реконструктивной операции может быть достигнута только временная компенсация местного кровотока.
Консервативная терапия
Консервативное лечение включает прием лекарственных препаратов, физиотерапевтические методы, лечебную физкультуру, иглорефлексотерапию. Для улучшения кровоснабжения мозга используют несколько групп лекарственных средств:
- Сосудорасширяющие средства (кавинтон, актовегин, цераксон).
- Нейропротекторы и ноотропы (пирацетам, глицин, пикамилон, мексидол), улучшающие обменные процессы в мозговой ткани.
- Бетагистин, эффективный при наличии головокружений.
- Антигипертензивные средства необходимы в случае повышения артериального давления: антагонисты кальция (амлодипин), бета-адреноблокаторы (бисопролол), ингибиторы АПФ - ангиотензинпревращающего фермента (лизиноприл).
- Профилактику образования тромбов осуществляют с помощью антиагрегантов (аспирин, пентоксифиллин, клопидогрель).
Из физиотерапевтических методов могут применяться:
- диадинамические токи;
- магнитотерапия;
- электрофорез с препаратами, обладающими сосудорасширяющим, обезболивающим действием.
- процедура магнитотерапии.
Одних таблеток мало. Важно изменить и образ жизни:
- Минимум животного жира, простых углеводов. Больше растительной пищи.
- Ограничение соли (до 7 граммов) и сахара.
- Адекватная физическая активность. В рамках дозволенного. Характер лучше уточнить у врача.
- Отказ от курения, спиртного.
- Адекватный сон. Не менее 8 часов за ночь.
- Избегание стрессов. Нервных перегрузок.
- Также не стоит резко поворачивать голову, изнурять себя физически.
Хирургическое лечение
Хирургическое вмешательство может осуществляться открытым способом или с помощью эндоваскулярного метода (через небольшие отверстия, без крупных разрезов).
Для восстановления кровотока применяют:
- Стентирование, при котором в место сужения сосуда вводят стент - каркас для расширения суженного участка. Такие стенты могут быть пропитаны лекарственными средствами.
- Ангиопластику, при которой в зону сужения вводят баллончик, который накачивают воздухом для расширения сосуда. Ангиопластика и стентирование могут дополнять друг друга.
- В тяжелых ситуациях проводят более сложную реконструктивную операцию: удаление деформированного участка и протезирование с помощью собственной вены пациента.
К сожалению, «традиционные» методы это все, чем официальная медицина может помочь пациенту. Однако, так называемые «центры альтернативной медицины», готовы порадовать пациента разнообразными процедурами. Здесь вам предложат в качестве «новой» терапии иглоукалывание, массаж, различные комплексы упражнений. Если вы решили опробовать новые методики, посоветуйтесь с лечащим врачом.
Последствия
Малый диаметр позвоночной артерии опасен только при сочетании с другими изменениями сосудов. В этих случаях может нарушаться питание всего головного мозга. Наиболее часто встречаются следующие сопутствующие состояния:
- атеросклероз сонных и позвоночных артерий;
- гипертоническая болезнь;
- незамкнутый Виллизиев круг (врожденная аномалия строения).
Все эти изменения сами по себе приводят к изменению кровообращения головного мозга и увеличивают риск инсульта. Сочетание с гипоплазией позвоночной артерии дополнительно повышает этот риск.
Основное последствие гипоплазии — инсульт. Без терапии при отрицательном сценарии его не избежать. Также возможна сосудистая деменция (слабоумие), падение качества жизни. В конечном итоге — инвалидность.
Гипоплазия позвоночной артерии с лева — результат врожденной аномалии. Это порок. Однако с ним можно долго и качественно жить с незначительными ограничениями. Важно своевременно обратиться к квалифицированному врачу и довериться ему.
Прогноз
Прогноз при патологии гипоплазия правой позвоночной артерии зависит от степени недоразвития, компенсаторных механизмов организма, сопутствующих патологий. При отсутствии симптомов ухудшения мозгового кровотока или минимальных проявлениях патологии прогноз можно считать условно благоприятным.
Гипоплазию относят к предрасполагающим факторам развития инсульта. Согласно статистике, 70% преходящих нарушений мозгового кровообращения и 30% инсультов связаны с нарушением кровотока в вертебробазилярной системе. Поэтому обнаружение аномалии требует принятия активных профилактических мер, особенно при наличии других факторов риска.
Наличие выраженных проявлений вертебробазилярной недостаточности значительно ухудшает прогноз. При недостаточной эффективности консервативной терапии улучшить ситуацию может только хирургическое лечение. Хорошие результаты получают при использовании эндоваскулярного метода, который может осуществляться даже у пациентов высокого «хирургического риска».
Профилактика
Так как точные причины заболевания пока что не выяснены, предупредить его практически невозможно. Чтобы снизить риск развития гипоплазии позвоночных артерий у плода, женщинам рекомендуется еще на стадии планирования беременности пройти все исследования (в частности, анализ на TORCH-инфекции) и при необходимости получить адекватное лечение.
В период вынашивания ребенка будущей матери следует отказаться от вредных привычек, вести здоровый образ жизни и по возможности устранить из своей жизни негативные факторы, которые могут оказать влияние на развитие эмбриона.
Гипоплазия позвоночных артерий - не смертельный приговор. Примерно 10% населения Земли живут долгие годы, даже не догадываясь о наличии этой патологии. Риск развития осложнений увеличивается только к старости, но при своевременном лечении, профилактике и соответствующем отношении к своему здоровью их вполне можно избежать.
Читайте также:
- Советы, как поддержать подругу, которая рассталась с парнем
- Псориаз: случай успешного лечения обострившейся болезни аппликациями из Нафталановой нефти
- Влияние содержимого кишечника на гены эпителиальных клеток кишечника
- Генерализованное поражение клеток при шоке. Ацидоз при шоке и некрозы тканей
- Осмотр кожи головы и шеи ЛОР-врачом