Синдром 47,XYY
Обновлено: 24.04.2024
Патологии половых хромосом связаны с нарушением их количества (т. е. анеуплоидии, например, моносомия X-хромосомы) или со структурными дефектами (например, такие геномные перестройки, как синдром дупликации гена MECP2). Частота врожденных хромосомных мутаций составляет как минимум 1:400.
Краткое содержание
Патологии половых хромосом могут быть обусловлены нарушением их количества (анеуплоидиями) или же структурными дефектами.
Наиболее распространенные анеуплоидии половых хромосом: 45,X (Синдром Тёрнера); 47,XXY (Синдром Клайнфельтера); 47,XYY; и 47,XXX. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует доле аномальных клеток.
Структурные патологии X- и Y-хромосом прежде всего включают изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Одним из примеров геномного расстройства является дупликация гена MECP2 у мужчин, выражающаяся в наличии мышечной гипотонии, тяжелой умственной отсталости, задержки речевого развития, нарушения глотания, частых респираторных инфекций, а также судорожных приступов (тонико-клонические судороги, не поддающихся лечению).
Аномалии числа хромосом (анеуплоидии)
Наиболее частыми анеуплоидиями половых хромосом являются 45,X (Синдром Шерешевского-Тернера); 47,XXY (Синдром Клайнфельтера); 47,XYY и 47,XXX с частотой возникновения приблизительно 1/2500, от 1/500 до 1/1000, от 1/900 до 1500 и 1/1000 соответственно. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом — 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует проценту аномальных клеток.
Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
Большинство пациентов с синдромом Шерешевского-Тёрнера имеют моносомию по Х-хромосоме, кариотип 45,X. Другие формы синдрома включают мозаицизм по хромосоме Х, например, 45,X/46,XX или 45,X/46,XY с частичной делецией Y-хромосомы. У некоторых пациентов имеется структурная аномалия второй X-хромосомы (например, изохромосомия длинного плеча X-хромосомы или делеция короткого плеча). Делеции, включающие в себя дистальную часть короткого плеча Y-хромосомы, также ассоциированы с фенотипом синдрома Тёрнера, поскольку в данном случае у пациентов отсутствуют так называемые анти-тёрнеровские гены (SHOX, RPSY4 и ZFY). Делеции короткого плеча X-хромосомы также связывают с фенотипом синдрома Тёрнера. В большинстве представляют собой единичные случаи.
Синдром Шерешевского-Тёрнера характеризуется низкорослостью и некоторыми из следующих проявлений: дисморфия лица, включающая низко посаженные уши, кожные складки на шее, щитообразная грудная клетка (широкая, с большим расстоянием между сосками), лимфедема, вальгусная деформация локтевого сустава, короткая четвертая пястная кость, гипоплазия ногтевых пластин, пигментные пятна и врожденные пороки сердца. Среди пороков сердца типичными и наиболее часто встречающимся являются дефекты сосудов и коарктация аорты. Вдобавок у пациентов, страдающих синдромом Тёрнера, развиваются полосковидные гонады, наблюдается нарушение овуляции и задержка полового развития. Также встречаются дефекты развития почек (подковообразная почка). Лимфедема нижних отделов конечностей может быть единственным клиническим признаком, наблюдаемым у новорожденных. Лица с синдромом Тёрнера, несущие генетический материал Y-хромосомы, имеют повышенный риск развития гонадобластомы.
47,XXY Синдром Клайнфельтера
Синдром Клайнфельтера является самой распространенной патологией числа половых хромосом, вызывающей первичный гипогонадизм. Кариотип 47,XXY является результатом нерасхождения половых хромосом и может быть как материнским, так и отцовским по происхождению. Большинство случаев болезни обнаруживается постнатально и диагностируется при определении причин бесплодия, выявлении гинекомастии, крипторхизма или же неврологических нарушений.
Рис. Нерасхождение половых хромосом

Новорожденные мальчики с кариотипом 47,XXY фенотипически нормальны, с физиологически нормальными мужскими наружными половыми органами и без какой-либо видимой дисморфии. Основные клинические проявления синдрома Клайнфельтера, включающие высокий рост, маленькие яички и бесплодие (азооспермия), становятся выраженными в постпубертатном периоде. У пациентов с синдромом Клайнфельтера повышен риск психических расстройств, расстройств аутистического характера и социальных проблем. У пациентов с диагностированным синдромом Клайнфельтера следует оценивать неврологический статус и направлять к эндокринологу.
47,XYY
Лица с кариотипом 47,XYY имеют высокий рост, у них может отмечаться умеренная задержка в двигательном и речевом развитии. Для многих из них требуется повышенное внимание к обучению, но, как правило, все они учатся в основных общеобразовательных школах. Половое развитие проходит нормально и большинство мальчиков фертильны. Из-за невыраженности фенотипа и отсутствия связанных с этим проблем со здоровьем, многие лица с кариотипом 47,XYY на протяжении всей их жизни остаются недиагностированными.
Ранее сообщалось, что у мужчин с 47,XYY повышена агрессия, что выражается в их агрессивном поведении. Однако последующие крупномасштабные совместные исследования европейских и американских генетиков показали, что статистика повышенной криминальной деятельности мужчин с XYY коррелировала с их низким социально-экономическим статусом по причине низкого значения IQ (около 10 баллов), что приводило к определенным трудностям с законом и, чаще, незначительным правонарушениям. У лиц с 47,XYY отмечаются более высокие показатели синдрома дефицита внимания и гиперактивности, а также расстройств аутистического характера. Таким пациентам рекомендуется оценка их нервно-психического развития, учитывая широкую распространённость трудностей в обучении и поведенческих проблем.
47,XXX
47,XXX (она же трисомия по X-хромосоме) является самой распространенной патологией половых хромосом у женщин. Трисомия по Х-хромосоме диагностируется внутриутробно в ходе генетического скрининга. У женщин с кариотипом 47,XXX нет повышенного риска развития плода с хромосомными аномалиями.
Обследование 155 женщин с кариотипом 47,XXX показало, что 62 процента из них были физически нормальными. Таким образом, для большинства лиц с кариотипом 47,XXX диагноз никогда не устанавливается. У женщин с 47,XXX отмечается высокий рост; (средняя длина окружности головы варьирует в пределах 25 - 35 процентиль, однако к подростковому возрасту для многих может достигать 80 процентиль). Половозрелость и фертильность чаще всего в норме, но может отмечаться преждевременное угасание функции яичников.
В следующем обследовании одиннадцати младенцев с кариотипом 47,XXX было показано, что коэффициент интеллекта девочек с рождения был на 15-20 баллов ниже, чем у их братьев. Поэтому рекомендуется отслеживать задержки в развитии и выявлять наличие психологических проблем в дальнейшем.
Другие заболевания
Сообщалось о более чем ста случаях кариотипа 49,XXXXY, по меньшей мере двадцати случаях 49,XXXXX и нескольких - 49,XYYYY. Прослеживается прямая зависимость между числом дополнительных половых хромосом и тяжестью фенотипических проявлений у пациентов. В исследовании тетра- и пентасомии половых хромосом сделан вывод о том, что полисомия по X-хромосоме связана с более тяжкими последствиями, чем полисомия по Y-хромосоме. Было показано, что уровень интеллекта IQ снижается на 10 пунктов с каждой лишней X-хромосомой от их нормального числа.
49,XXXXY Характерными клиническими чертами кариотипа XXXXY являются запавшая переносица с широким или приподнятым кончиком носа, широко расположенные глаза, веко-носовые складки, скелетные патологии (особенно лучелоктевой синостоз), врожденные сердечные заболевания, эндокринные расстройства и высокая степень гипогонадизма и гипогенитализма. Также обычным являются выраженная умственная отсталость и умеренная низкорослость. Хотя лиц с таким кариотипом часто относят к случаям синдрома Клайнфельтера, все характерные черты XXXXY довольно отчетливо указывают именно на данный фенотип.
49,XXXXX У женщин с кариотипом 49,XXXXX (пентасомия по X-хромосоме) всегда присутствует умственная отсталость. Другие проявления, такие как черпено-лицевые, сердечно-сосудистые и скелетные патологии довольно непостоянны. У пациентов, страдающих пентасомией по X-хромосоме, могут проявляться схожие черты с теми, что наблюдаются при синдроме Дауна. Лучелоктевой синостоз также часто выражен у пациентов с большим числом X-хромосом. Некоторые пациенты имеют мозаицизм 48,XXXX и 49,XXXXX.
Мозаицизм 45,X/46,XX
Это наиболее распространенный мозаицизм половых хромосом, который диагностируется при амниоцентезе и пренатальном кариотипировании. У лиц с данным типом мозаицизма имеются более легкие клинические черты синдрома Тёрнера. Многие женщины прошли половое созревание и смогли воспроизвести потомство.
Мозаицизм 45,X/46,XY
Мозаицизм с наличием 45,X/46,XY имеет широкий фенотипический спектр. Например, в ретроспективной серии 151 постнатально диагностированных случаев мозаицизма 45,X/46,XY, 42 % пациентов — девочки по фенотипу, с наличием типичного или нетипичного синдрома Тёрнера. Еще у 42 % наблюдались неопределённые наружные половые органы и асимметричные гонады (смешанный гонадный дисгенез), наконец, у 15% был мужской фенотип с неполной маскулинизацией. Таким образом, все случаи, диагностированные постнатально, были фенотипически патологичными. Напротив, среди 80 пренатально диагностированных случаев мозаицизма 45,X/46,XY 74 92,6% были нормальными по фенотипу мальчиками. Это может объяснить тот факт, что дети или взрослые с наличием мозаицизма, но нормальным фенотипом вряд ли стали бы обращаться за медицинской помощью (ошибка обращаемости).
Структурные аномалии хромосом
Структурные патологии включают, прежде всего, изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Изохромосома Xq
Изохромосома длинного плеча X-хромосомы, isoXq или i(Xq), при наличии которой короткое плечо (p) исключено (отсутствует/редуцировано) и заменено точной копией длинного плеча (q), — является наиболее распространенной аномалией половых хромосом.
Наличие структурной патологии не связывают с повышенным возрастным риском родителей. Изохромосомия 46,X,i(Xq) может быть выражением мозаицизма, когда в организме присутствуют две генетически разные клеточные популяции: нормальная - 46,XX и 45,X.
Изохромосомы Xq и Xy ассоциируют с синдромом Тёрнера, возможно, потому, что главный анти-тёрнеровский ген SHOX располагается на дистальной части коротких плеч X-и Y-хромосом (на псевдоаутосомных областях). Изохромосома Xq также выявляется у пациентов в одной из вариаций синдрома Клайнфельтера, 47,X,i(Xq),Y.
Делеция Xp22.11
Делеция Xp22.11 включает в себя ген PTCHD1. Сообщалось о выявлении в нескольких семьях с расстройствами аутистического характера, а также в трёх семьях с умственной отсталостью. Ген PTCHD1 является геном-кандидатом в отношении Х-сцепленной умственной отсталости, проявляющейся с аутизмом или без аутизма. Функция и роль данного гена неизвестны.
Делеция Xp22.3
Делеция данной области часто ассоциируется с синдромом микрофтальмии и линейных дефектов кожи (MLS) и является Х-сцепленным доминантным нарушением, то есть, летальным для мужчин и поэтому прослеживающимся только у женщин. Ген в данной области кодирует митохондриальную цитохром-c-синтазу (HCCS). Клиническое проявление MLS выражается наличием микрофтальмии и анофтальмии (одно- или двусторонней) и линейными дефектами кожи, в основном лица и шеи, которые со временем проходят. Структурные патологии головного мозга, задержка в развитии и приступы (припадки) тоже входят в состав клинической картины. Нарушения сердечной деятельности (как гипертоническая кардиомиопатия и аритмия), низкий рост, грыжа диафрагмы, ногтевая дистрофия, преаурикулярный свищ, потеря слуха, мочеполовые мальформации (пороки развития, неправильное формирование) также являются частыми клиническими явлениями.
Скрининговая оценка предусматривает офтальмологический и дерматологический осмотр, оценку общего развития, выполнение эхокардиограммы, магнитно-резонансной томографии мозга (МРТ) и электроэнцефалограммы (ЭЭГ).
Делеции Xp22 SHOX
Делеция Xp22 включает в себя ген SHOX, мутация которого является причиной идиопатического низкого роста. Ген SHOX находится в псевдоаутосомном регионе 1 X- и Y-хромосом. Этот ген считается ответственным за низкорослость при синдроме Тёрнера, а гаплонедостаточность данного гена вызывает дисхондростеоз Лери-Вейлля. Дисхондростеоз Лери-Вейлля характеризуется низким ростом, наиболее выражено проявляющимся у женщин, а также хроническим подвывихом кисти (деформацией костей запястья, деформация Маделунга). Гомозиготные делеции гена SHOX вызывают дисплазию Лангера, более тяжелую форму метафизарной дисплазии. Делеции гена SHOX легко обнаруживаются у пациентов с низким ростом, без каких-либо других специфических особенностей в строении их скелета. Более чем 60% перестроек SHOX - это делеции гена; при отсутствии делеций сравнительная геномная гибридизация с последующим секвенированием для выявления и установления точечных мутаций, является клиническим обследованием идиопатического низкого роста.
Делеции Xp11.22
Делеции региона Xp11.22 включают ген PHF8 (кодирует пальцевидный белок PHD8), мутации которого связывают с умственной отсталостью, наличием расщелины губы/неба, а также с расстройствами аутистического характера.
Мутации с делецией гена PHF8 ассоциированы с синдромом Х-сцепленной умственной отсталости, синдром Сидериус-Хамель (синдром Siderius-Hamel).
Дупликации Xp.22.31
Дупликации в локусе Xp.22.31 часто описываются в литературе. Было много дискуссий на тему того, является ли данная дупликация патогенетической или же доброкачественным явлением, учитывая трудности определения последствий вариации числа копий генов. Данная дупликация затрагивает ген стероидной сульфатазы. Как результат - генетический дефект, мутация в гене стероидной сульфатазы, что выражается в снижении её активности или отсутствие её синтеза. Делеция данного гена связана с Х-сцепленным ихтиозом у мужчин. Данная дупликация отмечается у пациентов с умственной отсталостью. Однако, она выявляется как у здоровых родственников этих пациентов, так и в основной популяции. Хотя дупликации данного гена могут и не иметь фенотипических проявлений, трипликации последовательно связывают с умственными расстройствами. FISH-диагностика позволяет в конечном счете дифференцировать дупликации от трипликации (распознать увеличение копийности гена).
Синдром дупликации ME2CP
Мутации в гене, кодирующем метил-связывающий-CpG терминальный белок 2 (ME2CP), расположенный в Xq28, ответственный за синдром Ретта. Дупликации данного региона имеет небольшое или вовсе не имеет фенотипического значения для женщин, вероятно, из-за инактивации патологической X-хромосомы. Мужчины с данной мутацией сильно ослаблены. Наличие дупликации клинически выражается в наличии выраженной мышечной гипотонии, тяжелой умственной отсталости, задержке речевого развития, нарушения глотания (трудностей приема пищи), частых респираторных инфекций и судорожных приступов вплоть до тонико-клонических, иногда не поддающихся лечению. Многие пациенты с наличием данной дупликации были с диагностированным аутизмом либо расстройством подобного типа. По аналогии с тем, что наблюдается в синдроме Ретта, пациенты с дупликацией ME2CP испытывают регресс развития. Вдобавок у них развивается атаксия, прогрессирующие мышечные спастичности нижней части тела часто приводят к потере способности передвигаться. Отмечались проблемы желудочно-кишечного тракта и сильные запоры. Дупликация часто затрагивает ген антагонист рецептора интерлейкина 1 (IRAK1), что может играть роль в появлении иммунных патологий, отмечаемой у данной группы пациентов. Прогноз неблагоприятен, и большинство мужчин с данной дупликацией умирают до 30 лет по причине вторичных респираторных инфекций. Трипликация данного региона проявляется еще более тяжелым фенотипом у мужчин.
Скрининговые обследования этих пациентов предполагают проведение ЭЭГ, оценку функции глотания, оценку гуморального и клеточного иммунитета. Лечение может включать лечение мышечной гипотонии и спастичности, речевую терапию (логопедию), использование гастрономической трубки (гастростома) в случае проблем с питанием, а также лечение респираторных инфекций.
Перевод материалов сайта UpTodate подготовлен специалистами Центра иммунологии и репродукции.
Синдром 47,XYY
Анеуплоидии половых хромосом: XYY синдром, XXY синдром (синдром Клайнфельтера), Х0 синдром (45Х0 синдром, синдром Тернера), ХХХ синдром
В большинстве случаев анеуплоидий половых хромосом (встречающихся приблизительно у 0,3% живых новорожденных) отмечается повышенный риск психиатрических отклонений. С другой стороны, анеуплоидии половых хромосом относительно часто встречаются среди детей, состоящих на учете психиатра. По результатам одного из исследований (Crandall et al., 1972) 1,6% детей, обратившихся к детскому психиатру, имели анеуплоидии половых хромосом.
Данные аномалии связаны с несколько повышенным риском задержки умственного развития. Например, по результатам исследования Ratcliffe et al. (1994) было продемонстрировано, что кариотип XXY выявлялся у 5,4 на 1000 фенотипических мальчиков, наблюдавшихся в учреждениях для лиц с умственной отсталостью, а коэффициент IQ у таких детей был несколько ниже, чем в контрольной группе.
Средний коэффициент IQ у девочек с тремя Х-хромосомами составил 86,8, у мальчиков с кариотипом XXY — 91,4, а у мальчиков с кариотипом XYY — 101,7; все показатели были значимо ниже, чем в контрольных группах (109,1 —у девочек и 115,8 — у мальчиков). Среди детей с дополнительной половой хромосомой также отмечалась меньшая окружность головы (даже при рождении), что отражает воздействие дополнительных половых хромосом на рост головного мозга в пре- и постнатальном периоде. Кариотип XXX был выявлен у 4,3 из 1000 фенотипических девочек, находящихся в лечебных учреждениях (Pennington et al., 1980).
Средний коэффициент IQ при кариотипе ХО (синдром Тернера) находится в пределах нормы, но многие девочки с данным кариотипом путают право-лево и имеют отклонения организации восприятия, их вербальный 1Q с определенной индивидуальной вариабельностью имеет склонность превышать IQ исполнения заданий (Temple и Carney, 1993).
Более серьезная умственная отсталость, очевидно, присуща пациентам со сложными аномалиями, к примеру, фенотипическим мужчинам с тремя и более Х-хромосомами (Zaleski et al., 1966) или с дупликацией обеих половых (X и Y) хромосом (Schlegel et al., 1965). Тем не менее, основные отклонения, встречающиеся у таких детей, включают сложности обучения и восприятия и поведенческие отклонения (Bender et al., 1983, Ratcliffe et al., 1994).
а) XYY синдром. У мальчиков с дополнительной Y-хромосомой (около 0,1% всего мужского населения) имеется значительно повышенный риск задержки и других отклонений речевого развития, приступов гнева и агрессивности. У таких пациентов также часто отмечается гипотония и раннее появление дефицита внимания с гиперактивностью. Приступы гнева часто встречаются в раннем детском возрасте. Распространена сниженная общительность, а риск аутизма повышен (Hagerman, 1989). Показатель IQ обычно в пределах нормы, но очень часто отмечаются трудности в обучении, а частота задержки умственного развития по сравнению с общей мужской популяцией повышена.
На основании как минимум одного объективного проспективного исследования предполагается небольшое повышение склонности к агрессивному поведению и проявления садизма в половой жизни во взрослом возрасте (Schiavi et al., 1988). Большинство пациентов имеет высокий рост и пропорциональное строение относительно размеров ног и головы. Пациенты обычно не менее чем на 13 см выше своего отца. Обследование на выявление кариотипа XYY необходимо у высокорослых мальчиков с отклонениями в поведении и трудностями в обучении.

Синдром Клайнфельтера
б) XXY синдром (синдром Клайнефельтера). У мальчиков с одной или более дополнительной Х-хромосомой (около 0,1-0,2% всех новорожденных мальчиков) диагностируется синдром Клайнфелтера (Lanfranco et al., 2004). Почти 2/3 случаев составляет кариотип 47XXY. Окружность головы, рост и вес при рождении ниже средних параметров. Приблизительно с возраста 2-4 лет отмечается увеличение скорости роста, особенно длины ног. Средний рост пациентов во взрослом возрасте приблизительно на 13 см превышает рост отца. Размер головы не компенсируется.
Дополнительная Х-хромосома ингибирует рост головного мозга во внутриутробном периоде. У мальчиков с синдромом Клайнфелтера обычно отмечается снижение вербального IQ, в то время как общий коэффициент IQ остается в пределах нормы (или слегка снижен); обычно коэффициент IQ варьирует от 60 до 130. Отмечается задержка речевого развития, и в большинстве случаев лечение по поводу отклонений развития речи начинается задолго до проведения хромосомной диагностики. Пациенты часто бывают неуклюжими и имеют отклонения типичные для детей с DAMP (дефицит внимания (Deficits in Attention), двигательного контроля (Motor control) и восприятия (Perception), иногда с тенденцией к гипоактивности.
Данные отклонения могут усиливаться за счет аномального строения тела и нервно-мышечных характеристик. Нередки серьезные проблемы с чтением и письмом, не зависящие от общего IQ. Изменения на МРТ в наиболее пораженных областях (лобной, височной и моторной зонах) и менее затронутой теменной области соответствуют выявляемым когнитивным и поведенческим отклонениям у пациентов с кариотипом XXY (Giedd et al., 2007).
Начиная со среднего детского возраста отмечается тенденция к удлинению ног, а размах рук часто превышает рост. Пенис может быть уменьшенного или нормального размера, яички почти всегда уменьшены, а продукция спермы нарушена. У многих пациентов отмечается увеличение молочных желез, а частота рака груди при данном заболевании выше, чем у здоровых мужчин. Часто пациенты характеризуются как застенчивые, неуверенные в себе. У большинства отмечаются легкие/умеренные проблемы социального взаимодействия и склонность избегать коллективной деятельности. У небольшого количества пациентов отмечается аутизм. Результаты одного проспективного исследования позволяют предположить, что взрослые пациенты с синдромом Клайнфелтера менее сексуально активны и более склонны к подчинению в сексуальной жизни, чем другие мужчины (Schiavi et al., 1988).
Лечение тестостероном следует начинать в препубертатном периоде под наблюдением эндокринолога, являющегося специалистом по синдрому Клайнфелтера. Интрацитоплазматическая инъекция спермы позволяет осуществлять детородную функцию, даже в случае, если в эякуляте сперматозоиды отсутствуют. Даже в случае стойкой азооспермии возможно выделение сперматозоидов из биптатов яичек и реализации, беременности и деторождения (Lanfranco et al., 2004).

Синдром Тернера
в) Х0 синдром (45Х0 синдром, синдром Тернера). Большинство случаев кариотипа 45X0 выявляется при рождении по характерному отеку тыла стоп и кистей и крыловидным складкам на шее. Кроме того, такие дети часто рождаются раньше срока с низкой массой тела и низким ростом. У девочек с данным синдромом частота психиатрических отклонений невысокая. Обычно 1Q в пределах нормы, но несколько снижен показатель исполнения заданий. Приблизительно у 10% пациентов отмечается определенный уровень задержки умственного развития. Зрительно-пространственные навыки часто развиты слабо, что может влиять на математические способности. По шкале детского интеллекта Векслера (WISC-R) отмечается плохое выполнение субтестов «композиции из кубиков» и «складывания фигур».
Многие пациенты с синдромом Тернера гиперактивны в раннем детском возрасте, но начиная с подросткового возраста склонны к гипоактивности (Swillen et al., 1993). Предполагается взаимосвязь синдрома Х0 с нервной анорексией (в частности, при мозаичном генотипе), но результаты систематического хромосомного исследования не подтверждают данного предположения (Rastam et al., 1991). После подросткового возраста пациенты характеризуются низкорослостью и инфантильным строением наружных и внутренних половых органов. Заместительная терапия эстрогенами необходима, но, тем не менее, не существует четких рекомендаций по поводу возраста начала лечения. Многие женщины с синдромом Тернера выходят замуж и во взрослом возрасте живут нормальной жизнью. Пациентки с синдромом Тернера, получившие Х-хромосому от матери, имеют больше проблем в социальном взаимодействии, чем женщины, получившие Х-хромосому от отца (Skuse et al., 1997).
г) ХХХ синдром. Девочки с кариотипом XXX составляют 0,1% новорожденных девочек. У таких детей имеется повышенный риск нарушений речи и обучения, IQ близок к нижней границе нормы, что приводит к необходимости применения специальных образовательных программ, тем не менее, нарушения чтения и письма соответствуют общему уровню IQ. Пациентки характеризуются застенчивостью, незрелостью и различными поведенческими проблемами (Linden et al., 1988). Характерен высокий рост и плохая координация движений. Данные отклонения в сочетании со странным поведением приводят к высокой частоте обращения в специализированные учреждения.
Мужской псевдогермафродитизм: признаки, фенотип
Кроме нарушения формирования яичка в эмбриональном периоде развития, причинами мужского псевдогермафродитизма у лиц с кариотипом 46,XY могут быть аномалии гонадотропинов, наследственные нарушения биосинтеза и метаболизма тестостерона и аномалии андрогенных клеток-мишеней.
Эти заболевания разнородны как генетически, так и клинически и в некоторых случаях могут оказаться мягкими проявлениями причин, лежащих в основе истинного гермафродитизма. Хотя при мужском псевдогермафродитизме гонады представлены исключительно яичками, происходит неполная маскулинизация внутренних половых протоков и наружных гениталий.
Дополнительно к мутациям или делециям любого из генов, задействованного в формировании и дифференцировке яичек, представленных ранее, существует множество форм нечувствительности к андрогенам, приводящих к мужскому псевдогермафродитизму.

Один из примеров — недостаточность стероид-5-а-редуктазы, фермента, отвечающего за преобразование мужского гормона тестостерона в его активную форму — дигидротестостерон. Это наследственное заболевание приводит к феминизации наружных половых органов у больных мальчиков. Хотя развитие яичек нормальное, половой член маленький и имеется короткое слепое влагалище. Могут возникать сложности с установлением пола.
Другое хорошо изученное заболевание — Х-сцепленный синдром, известный как синдром нечувствительности к андрогенам (прежде называвшийся тестикулярной феминизацией). При данном нарушении пораженные имеют мужской хромосомный пол (кариотип 46,XY) с внешне нормальными женскими наружными половыми органами, но слепым влагалищем и отсутствием матки и маточных труб.
Встречаемость нечувствительности к андрогенам около 1 на 20 000 новорожденных. Подмышечное и лобковое оволосение слабое или отсутствует. Старое название «тестикулярная феминизация» указывает на наличие яичек в пределах живота или паховых каналов, где их иногда ошибочно принимают за грыжу у детей, в остальном выглядящих как нормальные девочки. Таким образом, определение пола оказывается неверным, и психосексуальное развитие и сексуальные функции соответствуют нормальным женщинам (за исключением фертильности).
Хотя яички нормально выделяют андрогены, нечувствительность к ним конечных органов вызвана отсутствием андрогенных рецепторов в клетках-мишенях. Белок рецептора, определяемый нормальным аллелем в локусе Х-сцепленного рецептора андрогенов, образует комплекс с тестостероном и дигидротестостероном. Если комплекс не образуется, гормон не может стимулировать транскрипцию целевых генов, необходимую для дифференцировки в мужском направлении.
Молекулярный дефект определен в сотнях случаев и областей, от полной делеции гена до точечных мутаций в разных областях гена андрогенного рецептора.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Дополнительные Х и Y хромосомы
Генетическая информация содержится в каждой клетке в виде расположенных в ядре образований — хромосом. При этом у человека двойной (диплоидный) набор, то есть кариотип представлен 23 парами хромосом, а самих хромосом в норме 46.
Нормальная хромосомная формула может записываться, как 44+ХХ или 46(ХХ) — для женщин и 44+ХУ или 46(ХУ) — для мужчин. 44 хромосомы или правильнее 22 пары — аутосомы — они одинаковые для обоих полов, а 23-я пара называется половыми хромосомами, так как они определяют пол будущего ребенка.
Если 23-я пара — ХХ, то пол женский, если ХУ, то мужской.
Изменения общего числа хромосом, или нормального кариотипа называются анеуплоидиями. Хромосом при этом может быть больше или меньше нормы.
Виды анеуплоидий
Аутосомные
Изменения числа аутосом бывают по разным парам хромосом. Чаще всего встречаются трисомии по следующим парам хромосом, то есть ситуации, когда вместо двух хромосом — три. Трисомия по 21 паре — это синдром Дауна, трисомия по 18 паре — это синдром Эдвардса, трисомия по 13 паре — синдром Патау. Эти типы хромосомных патологий могут сочетаться с рождением живого плода, но со множественными тяжёлыми пороками развития, причём трисомии 18 и 13 отличаются очень слабой выживаемостью таких детей, а трисомия 21 несмотря на пороки развития позволяет человеку прожить достаточно длительный срок. Информация по основным трисомиям есть в соответствующих статьях на нашем сайте.
Вообще анеуплоидий по аутосомам значительно больше — они могут встречаться и по другим парам хромосом, значительно реже бывают тетрасомии или сочетания трисомий по нескольким парам одновременно, однако все они фатальны, и еще на самых ранних сроках беременности происходит самопроизвольное её прерывание.
Дополнительные Х и У хромосомы по 23 паре
Дополнительные хромосомы по 23 паре или дополнительные половые хромосомы формируют характерные клинические признаки, однако по степени своих проявлений дополнительные половые хромосомы сопровождаются значительно более мягкими дефектами. При этом, пусть очень редко, но могут встречаться тетра-, пента- и большее число хромосом, что приводит к усилению выраженности дефектов развития.
Ниже будут рассмотрены наиболее часто стречающиеся варианты дополнительных У и Х хромосом.
Синдром ХХУ — синдром Клайнфельтера
Кроме этого классического варианта есть другие более редкие сочетания с увеличением Х и У хромосом.
Данный синдром анеуплоидий по половым хромосомам характеризуется нарушением развития мужского организма в сторону его феминизации, то есть с преобладанием женских черт. Важно то, что до начала полового созревания в ряде случаев он может быть вообще не распознан, его можно лишь заподозрить по ряду косвенных признаков.
После наступления пубертатного возраста проявления становятся характерными — высокий рост, при этом женский тип фигуры с широким тазом и узкими плечами. Голос может оставаться практически без изменений — не мутирует, не становится мужским. Характерным является недоразвитие наружных половых органов и эректильная дисфункция. При этом отмечается оволосение по женскому типу. Отмечается более низкий уровень либидо у таких мужчин, очень часто возникает бесплодие ввиду нарушения развития сперматозоидов. В интеллектуальной сфере возможно некоторое отставание.
Синдром ХУУ — синдром «супермужчины»
Несмотря на название, никаких суперспособностей у таких людей нет, как впрочем и излишней агрессии, и чрезмерного потенциала в интимной сфере.
Основные черты — более низкий рост и более ранее начало полового созревания. Маскулинизация наступает раньше, нежели у сверстников, чаще встречается избыточное оволосение и ранее облысение. В интеллектуальной сфере отмечается незначительное отставание от популяции, но это связано с тем, что у таких детей с раннего детства повышенная отвлекаемость, лабильность внимания, неусидчивость, они труднее усваивают материал, именно со школы наблюдается указанное отставание. Может отмечаться некоторое снижение способности к зачатию. Хотя подавляющее большинство пациентов с синдромом ХУУ выявляется не клинически, а при различных генетических тестах, то есть «попутно»
Синдром ХХХ — синдром «суперженщины»
Аналогично предыдущему может быть «случайной находкой» при генетических исследованиях, внешне имеет минимальные проявления.
Из наиболее выраженных — некоторое снижение способности к вынашиванию, за счет большего процента самопроизвольных прерываний, могут наблюдаться минимальные проявления в репродуктивной и интеллектуальной сферах.
Дополнительные У и Х хромосомы с минимальными проявлениями
В данном случае речь идет о так называемом «мозаицизме». Это явление, когда определенная часть клеток организма содержит какой-либо вариант анеуплоидии, а остальные клитки описываются нормальной хромосомной формулой. Такая ситуация возникает, если хромосомная мутация возникла не при оплодотворении яйцеклетки, а на ранних стадиях деления зиготы. Чем позднее это происходит, тем меньшая часть клеток имеет нарушения кариотипа и тем меньше клинические проявления. Очень часто такой мозаицизм обнаруживается только при генетических исследованиях.
Диагностика различных типов анеуплоидий.
Единственным достоверным способом ранней внутриутробной диагностики хромосомных аномалий у плода является генетическое исследование. Подтверждение диагноза, заподозренного на основании выявленных при скрининге рисков, проводится с инвазивным забором материала, что требует чётко обоснованных показаний к проведению.
С помощью неинвазивного пренатального теста Пренетикс можно исследовать ДНК плода в венозной крови будущей матери начиная с 10-й недели беременности. Надёжность и специфичность метода позволяет отнести Пренетикс к скринингу экспертного уровня, значительно повышающему информативность традиционного скрининга первого триместра.
Приём в лаборатории - по предварительной записи
Неинвазивный пренатальный тест Пренетикс – надёжный и наиболее полно исследованный тест,
разработанный компанией Roche для скрининга хромосомных аномалий плода
по крови матери на сроке от 11 акушерских недель. Выполняется от 3-х рабочих дней.
Узнать цены
Синдром Якобcа - XYY
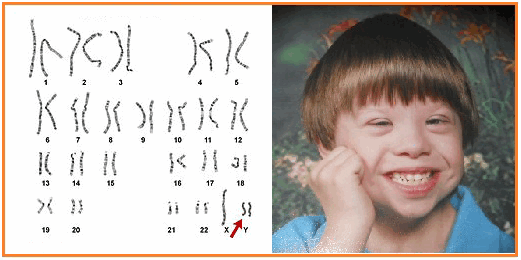
XYY-синдром — хромосомное заболевание, характерное только для мужчин. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Внешне мужчины с дополнительной Y-хромосомой обычно не имеют существенных отличий от нормальных, но могут иметь ряд особенностей.
Узнать с точностью 99% о риске синдрома Якобса и других хромосомных аномалий, а также пол плода, можно с 9 недель беременности, всего лишь сдав кровь из вены. Подробнее о НИПТ-тесте.
Признаки Синдрома Якобса
Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям. В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75 % мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими. Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы. Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения речи и чтения. Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы.
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с синдромом Якобса, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий. Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности). В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A. Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на синдром Якобса, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
Читайте также: